Degenerative Diseases of the Nervous System: Introduction
The adjective degenerative has no great appeal to the modern neurologist. It is also not an entirely satisfactory term medically, as it implies an inexplicable decline from a previous level of normalcy to a lower level of function—an ambiguous conceptualization of disease that satisfies neither a clinician nor a scientist. Moreover, it gives no hint as to the fundamental causation of a process and in all likelihood combines a number of mechanisms under 1 nondescript term. It would be tempting to attribute all progressive disease of the nervous system that are of unknown cause to degeneration. The problem is that many degenerative diseases of mundane type are caused in a proportion of cases by germ line genetic changes. All are currently called degenerative, but this nosology may be a transitional method of holding a place while awaiting more refined understanding. What is lacking at the moment is a precise subcellular mechanism for cellular loss; that is knowledge that a protein aggregates within or between cells is not equivalent to understanding the cause of an illness.
Gowers in 1902 suggested the term abiotrophy to encompass the degenerative diseases, by which he meant a lack of “vital endurance” of the affected neurons, resulting in their premature death. This concept embodies an unproven hypothesis—that aging and degenerative changes of cells are based on the same process. Understandably, contemporary neuropathologists are reluctant to attribute to simple aging the diverse processes of cellular diseases that are constantly being revealed by ultrastructural and molecular genetic techniques. It is increasingly evident that many of the diseases included in this category depend on genetic factors. Some appear in more than one member of the same family, in which case they may be properly designated as heredodegenerative. Even more diseases, not differing in any fundamental way from the heredodegenerative ones, occur sporadically, that is as isolated instances but still, genetic factors such as single nucleotide polymorphisms and copy number variations are often involved in pathogenesis.
Degeneration is nonetheless used as a clinical and pathologic term that refers to a process of neuronal, myelin, or tissue breakdown, the degradative products of which evoke a reaction of phagocytosis and cellular astrogliosis. What characterizes the degenerative disease as much as the loss of cells is the concentration of damage in functionally related cells, or systems; for example the cerebral cortex, motor system, extrapyramidal apparatus, or cerebellum, which are representative of the structures that are the targets of damage in this class of disease.
The basis of aging changes is also explainable at the neuronal level, but the nature of these alterations is not understood. A fundamental problem is the distinction of these aging deteriorations from degenerative disease. When a degenerative neurological disease appears in adult life, one must assume that the clinical presentation is modified to some extent by life-cycle phenomena—the patient’s function being a sum of both processes. However, their separation is of fundamental importance in diagnosis and therapeutics. One has to reconcile the fact that most degenerative diseases manifest themselves in later life, leading to the tentative conclusion that some aspect of the aging process is entwined with the cellular degenerations of disease. This creates a problem for the clinician, who may be inclined to attribute changes in a person’s function to aging alone rather than searching for a disease that may allow for treatment or for specific prognostication and counseling. Moreover, a long-standing uncertainty pertains to certain degenerative conditions such as Alzheimer disease, which becomes so prevalent in later age as to offer the possibility that the disease is an invariable aspect of aging rather than an acquired perturbation in cellular function. For most degenerative diseases of the nervous system, however, this inevitability of occurrence with aging is clearly not the case. For example, the proportional incidence of Alzheimer pathologic change decreases continuously from age 70 to age 100 according to Savva and colleagues. This polemic regarding aging and degenerative disease is irresolvable and exposes difficulties with meaning of the term “disease.” If the human being lived another 50 years beyond the current expectation, would all nervous structures show the changes of degenerative disease? The answer is probably “no,” as there are distinctive cellular and subcellular features of degenerative diseases that are different from the uncomplicated, programmed loss of cells that is due to aging.
Much new and essential information has been gained regarding the biologic derangements that lead to neuronal death and dysfunction as a result of investigating the inherited forms of degenerative diseases. The application of the techniques of molecular genetics to these diseases has given stunning results. Even when the hereditary form of a degenerative condition is rare in comparison to the sporadic type, general principles have been exposed that are common to the mechanisms of both forms of the disorder. This approach holds promise for effective treatment of what heretofore have been considered progressive and incurable diseases.
It has been proposed that all degenerative diseases be classified according to their genetic and molecular abnormalities. However, when one notes the diversity of pathologic change that may accompany a single, seemingly unitary gene abnormality or, reciprocally, the diversity of genetic defects that may underlie a single phenotype, this type of classification does not prove immediately helpful to the clinician. In other words, the practice of creating new disease categories to encompass all the molecular and pathologic changes associated with a particular type of neuronal degeneration offers no great advantage in practice. For example, certain diseases are unified by the deposition of proteins such as tau and have been termed “tauopathies,” “synucleinopathies,” “amyloidopathies,” and so forth. We endorse a more useful clinical approach that is based on an awareness of constellations of clinical features that relate to degeneration of neural systems. Until such time as the causation of the degenerative neurologic diseases is known, there must be a name and a place for a group of diseases that are united only by the common attribute of gradually progressive disintegration of a part or parts of the nervous system.
The diseases included in the degenerative category have 2 outstanding characteristics: (1) They affect specific parts or functional systems of the nervous system and (2) They begin insidiously, after a long period of normal nervous system function, and pursue a gradually progressive course. Frequently, it is impossible to assign a date of onset. The patient or the patient’s family may give a history of the abrupt appearance of disability, particularly if some injury, infection, surgical procedure, stroke, or other memorable event coincided with the initial symptoms. A skillfully taken history will reveal that there had been subtle symptoms for some time but had attracted little attention. Whether trauma or other stress can actually evoke or aggravate a degenerative disease is a question that cannot be answered with certainty; at present, evidence to this effect is largely anecdotal. Instead, these degenerative disease processes, by their very nature, appear to develop de novo, without relation to known antecedent events, and their symptomatic expressions are late events in the pathologic process, occurring only when the degree of neuronal loss exceeds the ability of a system to function at a clinically acceptable level. Irreversibility and steady progression of clinical manifestations when measured over periods of months or years is another feature common to the neurodegenerative conditions. However, several of these diseases sometimes display periods of relative stability.
Although most degenerative disease do not manifest expression in other members of the family, the familial occurrence of degenerative disease is of great importance both clinically and for scientific reasons as mentioned earlier, but such information is often difficult to obtain. The family may be small or widely scattered, so that the patient is unaware of the health of other members. The patient or the patient’s relatives may be reluctant to acknowledge that a neurologic disease has affected another family member. Furthermore, it may not be realized that an illness is hereditary if other members of the family have a much more or much less severe, or a different form of the disorder than the patient. Or paternity may be in question. Even without clear familial occurrence, the patient’s ethnicity may give clues to a propensity for certain diseases. Sometimes only the careful examination of other family members will disclose the presence of a hereditary disease. Also, it should be remembered that familial occurrence of a disease does not necessarily mean that it is inherited, but may indicate instead that more than one member of a family had been exposed to the same infectious or toxic agent.
Many symptoms of degenerative disease, while not currently curable, can be alleviated by skillful management. The physician’s interest and advice are invaluable to the patient and his family by way of providing support, perspective, and information. This accords with the highest calling of the physician’s abilities to relieve suffering.
Most of the degenerative diseases, as emphasized in the earlier general comments, are characterized by the selective involvement of anatomically and physiologically related systems of neurons. This feature is exemplified by amyotrophic lateral sclerosis (ALS), in which the pathologic process is virtually limited to motor neurons of the cerebral cortex, brainstem, and spinal cord, and by the progressive ataxias, in which only the Purkinje cells of the cerebellum are affected. Many other examples could be cited (e.g., Friedreich ataxia, Parkinson disease) in which discrete neuronal systems disintegrate, leaving others unscathed. Thus, these degenerative diseases had in the past been called system atrophies. The selective vulnerability of certain systems of neurons is not an exclusive property of the degenerative diseases; several different processes of known cause have similarly circumscribed effects on the nervous system. Contrariwise, in many degenerative diseases, the pathologic changes are somewhat less selective and eventually quite diffuse. Even then, there is an early tendency to involve special categories of neurons.
As one would expect of any pathologic process that is based on the slow wasting and loss of neurons, not only the cell bodies but also their dendrites, axons, and myelin sheaths disappear, unaccompanied by an intense tissue reaction or cellular response. The cerebrospinal fluid (CSF) shows little, if any, change, or at most a slight increase in protein content. Moreover, because these diseases invariably result in tissue loss, imaging examination shows either no change or only a volumetric reduction (atrophy) with a corresponding passive enlargement of the CSF compartments. These findings distinguish the neuronal atrophies from other large classes of progressive disease of the nervous system, namely, tumors, infections, and processes of inflammatory type.
At the cellular level, several processes characterize the death of individual cells. Among these mechanism is apoptosis, a term borrowed from embryology to specify the mechanisms that lead to neuronal degeneration. The original meaning of the term refers to a naturally occurring cell death during development that is driven by the expression of genes over a short period of time (i.e., “programmed” cell death), leaving no trace of a pathologic reaction. The process of neuronal degeneration is quite different in that it refers to a series of changes in mature neurons that occur over a protracted period of time, leading to cell death and often leaving a discrete glial scar, but not to regional tissue necrosis. In some models of degenerative disease, cell loss involves activation of specialized genes, although the time course and cellular morphology are not apoptotic in the original sense of the term. It is increasingly apparent that mechanisms other than programmed cell death will prove central to understanding the degenerative diseases, and that the clinical features of these conditions are manifest even before cellular destruction occurs. For example, interference with synaptic signaling and dysfunction of supporting glia cells are equally important to morphologic neuronal death.
It will become clear in the following discussion that the current theme in the study of degenerative diseases is that of aggregation within specific neurons of normal cellular proteins such as amyloid, tau, synuclein, ubiquitin, and huntingtin. In some cases, the protein is overproduced as a result of the simple fact of a triplication or overactivity of its corresponding gene. In other instances, enzymatic cleavage of a normal precursor protein yields a product with physical properties that lead to its aggregation (as happens with amyloid in Alzheimer disease) or, there may be failure of the normal mechanisms of protein removal, resulting in its excess accumulation. As mentioned above, this has resulted in the denomination of groups of diseases by the type of protein aggregate: tauopathy, synucleinopathy, etc. Even this is an uncertain or intermediate classification as it is not known in most cases if the protein is the cause or the result of cellular damage, and in any case, the fundamental mechanisms of cellular destruction are still being determined.
Another characteristic that has guided understanding of degenerative disease is the possible contiguous “spread” of protein aggregation from one to another region by synaptic connections. In some cases, this results in adjacent regions being affected sequentially and in others, circuits that are functionally integrated but not necessarily contiguous areas are affected. This geographic mechanism, proposed by Braak and Braak, conforms to certain pathologic observations such as the sequential appearance of synuclein in the olfactory system, thence in the Meissner-Auerbach plexus of the gut, followed by the vagus, to involvement of the vagal nuclei in the medulla, ascending trans-synaptically to the pons and midbrain nuclei. Whether this accounts for the selectivity of disease in areas such as the substantia nigra that is most affected in Parkinson disease, is not entirely known. In any case, the biologic and the physicochemical properties of these aggregated proteins have assumed great importance and the mechanisms by which they interfere with cellular function and potentially cause cell death are major areas of research in the degenerative diseases.
Clinical Classification
Because grouping of the degenerative diseases in terms of etiology is not entirely possible (except that a hereditary or genetic factor can be recognized in some), we resort for practical purposes to a division based on the presenting clinical syndromes and their pathologic anatomy. Although this is the most elementary mode of classification of naturally occurring phenomena, it is a necessary prelude to diagnosis and scientific study and preferable to a purely genetic or molecular classification. It is certainly an improvement on a haphazard listing of diseases by the names of the neurologists or neuropathologists who first described them. For reasons given in the introduction to this chapter, this approach remains the most effective in analyzing the problem presented by an individual patient. The main clinical categories are as follows:
Syndrome of progressive dementia, other neurologic signs absent or inconspicuous
Alzheimer disease
Some cases of Lewy-body disease
Frontotemporal dementias—Pick disease, including behavioral variant, primary progressive aphasias (several types)
Posterior cortical atrophy (visuospatial dementia)
Syndrome of progressive dementia in combination with other neurologic abnormalities
Huntington disease (chorea)
Lewy-body disease (parkinsonian features)
Some cases of Parkinson disease
Corticobasal ganglionic degeneration (rigidity, dystonia)
Cortical-striatal-spinal degeneration (Jakob disease)
Dementia-Parkinson-amyotrophic lateral sclerosis complex
Cerebrocerebellar degeneration
Familial dementia with spastic paraparesis, amyotrophy, or myoclonus
Polyglucosan body disease (neuropathy)
Frontotemporal dementia with parkinsonism or ALS
Syndrome of disordered posture and movement
Parkinson disease
Multiple system atrophy, MSA-P (striatonigral degeneration, Shy-Drager syndrome)
Progressive supranuclear palsy
Dystonia musculorum deformans
Huntington disease (chorea)
Acanthocytosis with chorea
Corticobasal ganglionic degeneration
Lewy-body disease
Restricted dystonias, including spasmodic torticollis and Meige syndrome
Essential tremor
Syndrome of progressive ataxia
Spinocerebellar ataxias
Friedreich ataxia
Non-Friedreich, early-onset ataxia (with retained reflexes, tremor, hypogonadism, myoclonus, and other disorders)
Cerebellar cortical ataxias
Holmes type of familial pure cerebellar-olivary atrophy
Late-onset cerebellar atrophy
Complicated hereditary and sporadic cerebellar ataxias (later-onset ataxia with brainstem and other neurologic disorders)
Multiple system atrophies (MSA-C) including olivopontocerebellar degenerations (OPCA)
Dentatorubral degeneration (Ramsay Hunt type)
Dentatorubropallidoluysian atrophy (DRPLA)
Machado-Joseph (Azorean) disease; SCA-3
Other complicated late-onset, autosomal dominant ataxias with pigmentary retinopathy, ophthalmoplegia, slow eye movements, polyneuropathy, optic atrophy, deafness, extrapyramidal features, and dementia
Syndrome of slowly developing muscular weakness and atrophy
Motor disorders with amyotrophy
Amyotrophic lateral sclerosis
Progressive spinal muscular atrophy
Progressive bulbar palsy
Kennedy syndrome and other hereditary forms of progressive muscular atrophy and spastic paraplegia
Motor neuron disease with frontotemporal dementia
Spastic paraplegia without amyotrophy
Primary lateral sclerosis
Hereditary spastic paraplegia (Strümpell-Lorrain)
Sensory and sensorimotor disorders (neuropathies; see Chap. 46)
Hereditary sensorimotor neuropathies—peroneal muscular atrophy (Charcot-Marie-Tooth); hypertrophic interstitial polyneuropathy (Dejerine-Sottas)
Pure or predominantly sensory or motor neuropathic
Riley-Day autonomic degeneration
Syndrome of progressive blindness with or without other neurologic disorders (see Chap. 13)
Pigmentary degeneration of retina (retinitis pigmentosa)
Stargardt disease
Age-related macular degeneration (ARMD)
Syndromes characterized by degenerative neurosensory deafness (see Chap. 15)
Pure neurosensory deafness
Hereditary hearing loss with retinal diseases
Hereditary hearing loss with system atrophies of the nervous system
Diseases Characterized Mainly by Progressive Dementia
This is the most common and important degenerative disease of the brain, having an immense societal impact. Some aspects of the intellectual deterioration that characterize this disease were described in Chap. 21, under “The Neurology of Dementia,” and the still ambiguous relationship of this disease to the aging process is mentioned above and in Chap. 29. There it was pointed out that some degree of shrinkage in size and weight of the brain, that is “atrophy,” is an inevitable accompaniment of advancing age, but that these changes alone are of relatively slight clinical significance and uncertain structural basis (e.g., whether the loss of brain weight aging is the result of a simple depletion of neurons). By contrast, severe degrees of diffuse cerebral atrophy that evolve over a few years are associated with dementia, and the underlying pathologic changes in these cases most often prove to be those of Alzheimer disease. As also commented on in Chap. 29, the rate of cerebral atrophy, specifically of the hippocampus and medial parts of the temporal lobes, is accelerated in the early stages of Alzheimer disease, and longitudinal studies by magnetic resonance imaging can identify individuals who will subsequently develop the disease (Rusinick). Nevertheless, there is not a continuous increase in the deposition of plaques and tangles, the pathologic markers of Alzheimer disease, with increasing age. Therefore, Alzheimer changes are not an inevitable result of aging.
The now outdated practice of giving Alzheimer disease and senile dementia the status of separate diseases is attributable to the relatively young age (51 years) of the patient originally studied by Alois Alzheimer in 1907. Such a division is no longer tenable, as the 2 conditions, except for their age of onset, are clinically and pathologically indistinguishable. It is probably useful to consider as related but separable, the several heredofamilial forms of Alzheimer disease discussed below.
Although Alzheimer disease has been described at every period of adult life, the majority of patients are in their sixties or older; a relatively small number have been in their late fifties or younger. It is one of the most frequent mental illnesses, making up a large proportion of persons in assisted living and skilled nursing facilities. The incidence of clinically diagnosed Alzheimer disease is similar throughout the world, and it increases with age, approximating 3 new cases yearly per 100,000 persons younger than age 60 years and a staggering 125 new cases per 100,000 of those older than age 60 years. The prevalence of the disease per 100,000 population is near 300 in the group aged 60 to 69 years; it is 3,200 in the 70- to 79-year-old group and 10,800 in those older than age 80. In the year 2008, there were estimated to be more than 2 million persons with Alzheimer disease in the United States. (It should be borne in mind that these are not pathologically proven cases and, while probably correct as an approximation, are likely combined with other diseases.) Prevalence rates, which depend also on overall mortality, are 3 times higher in women, although the incidence of new cases is only slightly disproportionate in women. The survival of patients with Alzheimer disease is reduced to half the expected rate, mainly because of respiratory and cardiovascular causes and inanition, but also for other reasons that are not entirely clear.
Several putative epidemiologic risk factors for Alzheimer disease, such as birth order, mother’s age at birth, and a family history of Down syndrome seem marginal at best and in some instances may be a result of selection bias. Depression and possibly head injuries do seem to confer a somewhat increased risk later in life. Whether low educational attainment is a risk factor for the development of Alzheimer disease or, conversely, whether cognitively demanding occupations or higher intelligence protects against dementia is still under discussion. Provocative data indicating that inherent intellectual endowment is important were presented in Chap. 21 (Katzman; Cobb et al). Finally, associations between diabetes or hyperglycemia and dementia, in general, have emerged from epidemiologic studies, for example, one reported by Crane and coworkers, but the ostensible mechanism by which this confers risk has not been established. In their report, a higher than average glucose level over the preceding 5 years conferred a slightly increased risk of dementia but not necessarily of Alzheimer disease.
The familial occurrence of Alzheimer disease has been well established. In less than 1 percent of such cases there is a dominant inheritance pattern with a high degree of penetrance and appearance of disease at a younger age (Nee et al; Goudsmit et al; see further). Reports of substantial familial aggregations of dementia without a specific pattern of inheritance also suggest the operation of more than one genetic factor. Many studies have documented an increase in the risk of ostensibly sporadic Alzheimer disease among first-degree relatives of patients with this disorder. Again, this risk is disproportionately greater in females, adding to the evidence that women in general are at slightly higher risk for Alzheimer disease (Silverman et al). Li and coworkers have provided evidence that patients with an earlier age of onset of Alzheimer disease (before age 70 years) are more likely to have relatives with the disease than are patients with later onset. Genetic studies are difficult to carry out because the disease does not appear at the same age in a given proband. Even in identical twins, the disease may develop at the age of 60 years in one of the pair and at 80 years in the other. Death from other causes may prevent its detection. The other genetic contributions to the occurrence of Alzheimer disease are discussed extensively further on.
The onset of mental changes is usually so insidious that neither the family nor the patient can date the time of its beginning and most patients come to attention months or years after the decline began. Occasionally, however, the process becomes manifest by an unusual degree of confusion in relation to a febrile illness, an operation, mild head injury, or the institution of a new medication. Other patients have as their initial complaints dizziness, mental fogginess, nondescript headaches, or other vaguely expressed and changeable somatic symptoms.
The gradual development of forgetfulness is the major symptom. Small day-to-day happenings are not remembered. Seldom-used names become particularly elusive. Little-used words from an earlier period of life also tend to be lost. Appointments are forgotten and possessions misplaced. Questions are repeated again and again, the patient having forgotten what was just discussed. It is said that remote memories are preserved and recent ones lost (the Ribot law of memory), but this is only relatively true and it is difficult to check the accuracy of distant personal memories. For example, Albert and associates, who tested Alzheimer patients’ recognition of dated political events and pictures of prominent people past and present, found that some degree of memory loss extends to all previous decades of the person’s life (neuropsychologic testing is discussed further on).
Once the memory disorder has become pronounced in the prototypic disorder, other failures in cerebral function become increasingly apparent. The patient’s speech is halting because of failure to access the needed word. The same difficulty interrupts writing. Vocabulary becomes restricted, and expressive language becomes stereotyped and inflexible. Comprehension of spoken words seems at first to be preserved, until it is observed that the patient does not carry out a complicated request; even then it is uncertain whether the request was not understood because of inattention or because it was forgotten. Almost imperceptible at first, these disturbances of language become increasingly apparent as the disease progresses. The range of vocabulary and the accuracy of spelling are reduced. Finally, after many years of illness, there is a failure to speak in full sentences; the finding of words requires a continuous search; and little that is said or written is fully comprehended. There is a tendency to repeat a question before answering it, and later there may be a rather dramatic repetition of every spoken phrase (echolalia). The deterioration of verbal skills has by then progressed beyond a groping for names and common nouns to an obvious anomic aphasia. Other elements of receptive and executive aphasia are later added, but discrete aphasias of the Broca or Wernicke type are characteristically lacking. In general, there is a paucity of speech and a quantitative reduction in mentation.
Skill in arithmetic suffers a similar deterioration. Faults in balancing the checkbook, mistakes in figuring the price of items and in making the correct change; all these and others progress to a point where the patient can no longer carry out the simplest calculations (acalculia or dyscalculia).
In some patients, visuospatial orientation becomes defective. The car cannot be parked; the arms do not find the correct sleeves of the jacket or shirt; the corners of the tablecloth cannot be oriented with the corners of the table; the patient turns in the wrong direction on the way home or becomes lost. The route from one place to another cannot be described, nor can given directions be understood. As this state worsens, the simplest of geometric forms and patterns cannot be copied.
Late in the course of the illness, the patient forgets how to use common objects and tools while retaining the necessary motor power and coordination for these activities. The razor is no longer correctly applied to the face; the latch of the door cannot be unfastened; and eating utensils are used awkwardly. Finally, only the most habitual and virtually automatic actions are preserved. Tests of commanded and demonstrated actions cannot be executed or imitated. Ideational and ideomotor apraxia are the terms applied to the advanced forms of this motor incapacity as described in Chaps. 3 and 22.
As these many amnesic, aphasic, agnostic, and apraxic deficits declare themselves, the patient at first seems unchanged in overall motility, behavior, temperament, and conduct. Social graces, whatever they were, are retained in the initial phase of the illness, but troublesome alterations may gradually appear in this sphere as well. Imprudent business deals may be made. Restlessness and agitation or their opposites—inertia and placidity—become evident. Dressing, shaving, and bathing are neglected. Anxieties and phobias, particularly fear of being left alone, may emerge. A disturbance of the normal day and night sleep patterns is prominent in some patients. A poorly organized paranoid delusional state, sometimes with hallucinations, may become manifest. The patient may suspect his elderly wife of having an illicit relationship or his children of stealing his possessions. A stable marriage may be disrupted by the patient’s infatuation with a younger person or by sexual indiscretions, which may astonish the community. The patient’s affect coarsens; he is more egocentric and indifferent to the feelings and reactions of others. A gluttonous appetite sometimes develops, but more often eating is neglected, resulting in gradual weight loss. Later, grasping and sucking reflexes and other signs of frontal lobe disorder are readily elicited (Neary et al), sphincteric continence fails, and the patient sinks into a state of relative akinesia and mutism, as described in Chap. 21.
Difficulty in locomotion, a kind of unsteadiness with shortened steps but with only slight motor weakness and rigidity, frequently supervenes. Elements of parkinsonian akinesia and rigidity and a fine tremor can be perceived in patients with advanced stages of the disease. Ultimately, the patient loses the ability to stand and walk, being forced to lie inert in bed and having to be fed and bathed, the legs curled into a fixed posture of paraplegia in flexion (in essence, a persistent vegetative state).
The symptomatic course of this illness is quite variable but usually extends over a period of 5 or more years, but judging from pathology studies, the pathologic course has a much longer asymptomatic duration. This concept of a preclinical stage is supported by the detailed studies of Linn and colleagues, who found that a lengthy period (7 years or more) of stepwise decline in memory and attention span preceded the clinical diagnosis. In the dominantly inherited forms of disease, careful studies of biomarkers in the spinal fluid and by imaging show that changes occur 15 years or longer before the clinical manifestations are apparent (Bateman et al). Throughout this period, corticospinal and corticosensory functions, visual acuity, ocular movements, and visual fields remain intact. If there is hemiplegia, homonymous hemianopia, and the like, either the diagnosis of Alzheimer disease is incorrect or the disease has been complicated by a stroke, tumor, or subdural hematoma. Exceptions to this statement are rare. The tendon reflexes are little altered and the plantar reflexes almost always remain flexor. There is no sensory or cerebellar ataxia. Convulsions are rare until late in the illness, when up to 5 percent of patients reportedly have infrequent seizures. Occasionally, widespread myoclonic jerks or mild choreoathetotic movements are observed late in the illness. Eventually, with the patient in a bedfast state, an intercurrent infection such as aspiration pneumonia or some other disease mercifully terminates life.
The sequence of neurologic disabilities may not follow this described order and one or another deficit may take precedence, presumably because the disease process, after becoming manifest in the memory cortex of the temporal lobes, affects a particular part of the associative cortex earlier or more severely in one patient than in another. This allows a relatively restricted deficit to become the source of early medical complaint, long before the full syndrome of dementia has declared itself.
There are at least 4 limited deficits that may represent the opening features of Alzheimer disease but each of which alone may be mild enough to qualify as mild cognitive impairment (MCI). According to Petersen, who developed this concept, the MCI syndrome is defined by the presence of cognitive difficulties in one or all spheres that are not severe enough to interfere with daily life.
The early presentation of Alzheimer disease may manifest mainly as one of the following syndromes with the first, memory dysfunction being the most common and, even as other aspects of the disease advance, it tends to remain the most prominent.
Amnesia The early stages of Alzheimer disease are usually dominated by a disproportionate failure of episodic (autobiographical) memory, with integrity of other cognitive abilities. This may be the sole difficulty for many years. In such patients, immediate memory (essentially a measure of attention), tested by the capacity to repeat a series of numbers or words, is intact; it is the short-and long-term (retentive) memory that fails. Memory may become impaired but as a business executive, for example, the individual may continue to make acceptable decisions if the work uses long-established habit patterns and practices.
Dysnomia The forgetting of words, especially proper names, may first bring the patient to a neurologist. Later the difficulty involves common nouns and progresses to the point where fluency of speech is seriously impaired. Every sentence is broken by a pause and search for the wanted word; if the desired word is not found, a circumlocution is substituted or the sentence is left unfinished. When the patient is given a choice of words, including the one that was missed, there may be a failure of recognition. Repetition of the spoken words of others, at first flawless, later brings out a lesser degree of the same difficulty. The defect in naming is evident with even simple tests, for example, asking the patient to generate a list of farm animals or car brands—a test that may elicit only 3 or 4 responses. A more extensive examination entails asking the patient to name as many items as possible in each of 3 categories in 1 min—vegetables, tools, and clothing. Alzheimer patients fall well below a score of 50 items.
Visuospatial disorientation Parietooccipital functions are sometimes deranged in the course of Alzheimer disease and in a few cases may fail while other functions are relatively preserved. When it occurs in a pure form it is termed posterior cortical atrophy, as discussed in a later section (see Renner et al). As remarked above and in Chap. 22, prosopagnosia (impaired facial recognition), losing one’s way in familiar surroundings or inability to interpret a road map, to distinguish right from left, or to park or garage a car, and difficulty in setting the table or dressing are all manifestations of a special failure to orient the schema of one’s body with that of surrounding space. Exceptionally, there is a neglect of stimuli in one visual field. In the late states, some of these patients develop the Balint syndrome or Gerstmann syndrome (Tang-Wai et al; McMonagle et al).
Paranoia and personality changes Occasionally, at some point in the development of Alzheimer dementia, paranoia or bizarre behavior occasionally assumes prominence. This may appear before the more obvious memory or language defects announce themselves. The patient becomes convinced that relatives are stealing his possessions or that an elderly and even infirm spouse is guilty of infidelity. He may hide his belongings, even relatively worthless ones, and go about spying on family members. Hostilities arise, and wills may be altered irrationally. Many of these patients are constantly worried, tense, and agitated. Of course, paranoid delusions may be part of a depressive psychosis and of other dementias, but most of the elderly patients in whom paranoia is the presenting problem, seem not to be depressed, and their cognitive functions are for a time relatively well preserved. Social indiscretions, rejection of old friends, embarking on imprudent financial ventures, or an amorous pursuit that is out of character are examples of these types of behavioral change.
Executive dysfunction This may be the most disabling of the main aspects of the disease and when it appears early on, is not specific to Alzheimer dementia as it is a component of several other processes that affect the frontal lobes. These patients display early difficulties in coordinating and planning tasks and following complex conversations or instructions. They may become disinclined to participate in social activities and become withdrawn or quieter than usual. As the problem advances, simpler and formerly automatic actions such as driving become problematic for the patient; the degree of insight varies. Some are able to express that they feel “confused’ but more often, it is the family that brings these changes to attention.
If one of the foregoing restricted deficits remains uncomplicated over a long period, one is justified in suspecting a cause other than Alzheimer disease, such as one of the lobar atrophies such as frontotemporal dementia (see further on), Binswanger disease, hydrocephalus, or embolic infarctions of the temporal or parietal lobes. Each of the restricted clinical disorders described above is only relatively pure. Careful testing of mental function—and this is of diagnostic importance—frequently discloses subtle abnormalities in several cognitive spheres. Initially, most patients have a disproportionate disorder of the temporoparietal cortices, reflected by an earlier impairment on the performance parts of the Wechsler Adult Intelligence Scale. Within a year or two, the more generalized aspects of mental deterioration become apparent, and the aphasic–agnosic–apraxic aspects of the syndrome become increasingly prominent. Although it is true that most patients with Alzheimer disease walk normally until relatively late in their illness, infrequently a short-stepped gait and imbalance draw attention to the disease and worsen slowly for several years before cognitive manifestations become evident. The general decrepitude in appearance that accompanies the middle and late stages of the disease in many patients is commented on in Chap. 21.
For research purposes and to establish certain inclusive and exclusive criteria for the diagnosis of Alzheimer disease, a working group of the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Diseases Association (ADRDA) had proposed the following criteria: (1) dementia defined by clinical examination, the Mini-Mental Scale (see Table 21-6), the Blessed Dementia Scale, or similar mental status examination; (2) patient older than age 40 years; (3) deficits in 2 or more areas of cognition and progressive worsening of memory and other cognitive functions, such as language, perception, and motor skills (praxis); (4) absence of disturbed consciousness; and (5) exclusion of other brain diseases (McKhann et al, 1984; Tierney et al, 1988). These criteria have essentially been reaffirmed by more recent consensus panels (see McKhann et al, 2011). Using these measures, the correct diagnosis is achieved in more than 85 percent of patients, but this is not surprising given that Alzheimer disease is overwhelmingly the most common cause of adult dementia. Most cases are identifiable without resorting to restrictive lists such as these, especially if the patient is observed serially over a period of months or years. There is strong interest in the addition of biomarkers to the diagnostic criteria for the disease but these have not reached the point of general clinical utility and the diagnosis remains predominantly a clinical one, aided by imaging and other tests.
In the advanced stages of the disease, the brain presents a diffusely atrophied appearance and its weight is usually reduced by 20 percent or more. Cerebral convolutions are narrowed and sulci are widened. The third and lateral ventricles are symmetrically enlarged to varying degrees. Usually, the atrophic process involves the frontal, temporal, and parietal lobes, but cases vary considerably. The extreme atrophy of the hippocampus, the most prominent finding visible on MRI (mainly coronal images), is diagnostic in the proper clinical circumstances.
Microscopically, there is widespread loss of nerve cells. Early in the disease this is most pronounced in layer II of the entorhinal cortex. In addition to marked neuronal loss in the hippocampus, adjacent parts of the medial temporal cortex—namely, the parahippocampal gyri and subiculum—are affected. The anterior nuclei of the thalamus, septal nuclei, and diagonal band of Broca, amygdala, and particular brainstem parts of the monoaminergic systems are also depleted. The cholinergic neurons of the nucleus basalis of Meynert (the substantia innominata) and locus ceruleus are also reduced in number, a finding that has aroused great interest because of its putative role of the former in memory function (see below). In the cerebral cortex, the cell loss predominantly affects the large pyramidal neurons. Residual neurons are observed to have lost volume and ribonucleoprotein; their dendrites are diminished and crowd one another owing to the loss of synapses and neuropil. Astrocytic hypertrophy (more than proliferation) is in evidence as a compensatory or reparative process, most prominent in layers III and V.
Moreover, 3 microscopic changes give this disease its distinctive character: (1) The presence within the nerve cell cytoplasm of thick, fiber-like strands of silver-staining material, also in the form of loops, coils, or tangled masses (Alzheimer neurofibrillary changes or “tangles”) (Fig. 39-1). These strands are composed of a hyperphosphorylated form of the microtubular protein, tau, and appear as pairs of helical filaments when studied ultrastructurally. (2) Spherical deposits of amorphous material scattered throughout the cerebral cortex and easily seen with periodic acid-Schiff (PAS); the core of the aggregates is the protein amyloid, surrounded by degenerating nerve terminals (neuritic plaques) that stains with silver. Amyloid is also scattered throughout the cerebral cortex in a nascent “diffuse” form, without organization or core formation and then is appreciated mainly by immunohistochemical methods, as well as deposition in the walls of small blood vessels near the plaques, so-called congophilic angiopathy. (3) Granulovacuolar degeneration of neurons, most evident in the pyramidal layer of the hippocampus. This last change is least important in diagnosis but there is uncertainty regarding its nature; it had been thought to be simply a reactive process but recent studies suggest it reflects a defect in phagocytosis of degraded proteins.
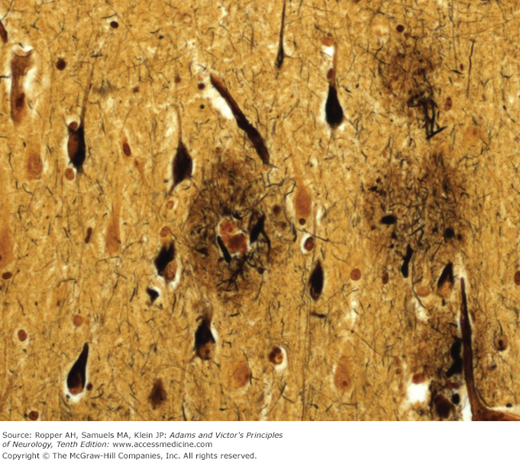
Neuritic plaques and neurofibrillary changes are found in all the association areas of the cerebral cortex, but it is the neurofibrillary tangles and quantitative neuronal loss, not the amyloid plaques, that correlate best with the severity of the dementia (Arriagada et al). If any part of the brain is disproportionately affected, it is the hippocampus, particularly the CA1 and CA2 zones (of Lorente de Nó) and the entorhinal cortex, subiculum, and amygdala. These parts have abundant connections with other parts of the temporal lobe cortex and dentate gyrus of the hippocampus and undoubtedly account for the amnesic component of the dementia. The associative regions of the parietal lobes are another favored site. Only a few tangles and plaques are found in the hypothalamus, thalamus, periaqueductal region, pontine tegmentum, and granule-cell layer of the cerebellum.
Experienced neuropathologists recognize a form of Alzheimer disease, particularly in older patients (75 years), in which there are senile plaques but few or no neuronal tangles (about 20 percent of 150 cases reported by Joachim et al). Increasingly, other pathologic changes are being appreciated in Alzheimer cases with fewer plaques and tangles than anticipated for the degree of dementia; Lewy bodies in particular are found by sophisticated techniques. Another problem for the neuropathologist is to distinguish between the normal-aged brain and that of Alzheimer disease. It is not unusual to find a scattering of senile plaques in individuals who were ostensibly mentally normal during life. Anderson and Hubbard studied 27 demented individuals aged 64 to 92 years and 20 age-matched nondemented controls. In the former, 3 to 38 percent of the hippocampal neurons contained neurofibrillary tangles; in all but 2 of the controls, the number of hippocampal neurons with tangles fell below 2.5 percent. Moreover, an increased number of tangles in the aged are associated with mild cognitive impairment and a higher likelihood of progression to Alzheimer disease.
Many demented individuals with clinical features of Alzheimer disease have sufficient neuronal loss and Lewy bodies in cortex and the substantia nigra to justify a diagnosis on histopathologic grounds of Parkinson disease (see further on). Leverenz and Sumi found that 25 percent of their Alzheimer patients showed the pathologic (and clinical) changes of Parkinson disease, a much higher incidence than can be attributed to chance. Similarly, of 11 patients with progressive supranuclear palsy (also discussed further on) reported by Gearing and coworkers, 10 were demented and 5 had the neuropathologic features of Alzheimer disease. These mixed cases present problems not only of classification but also in understanding the neurobiology of these degenerative diseases. This subject is discussed further in the section on Parkinson disease.
It is of historical interest that Alzheimer was not the first to describe plaques, one of the hallmarks of the pathologic state. Miliary lesions (Herdchen) had been observed in senile brains by Blocq and Marinesco in 1892 and were named senile plaques by Simchowicz in 1910. In 1907, Alzheimer described the case of a 51-year-old woman who died after a 5-year illness characterized by progressive dementia. Throughout the cerebral cortex he found the characteristic plaques, but he also noted, thanks to the use of Bielschowsky’s newly devised silver impregnation method, a clumping and distortion of fibrils in the neuronal cytoplasm, the neurofibrillary change that now, appropriately, carries Alzheimer’s name.
Analyses of the plaques and neuronal fibrillary changes have been undertaken in an attempt to elucidate the mechanism of Alzheimer disease, but so far, to little avail. Several histologic techniques assist in this endeavor, including refined methods for silver impregnation that stain both amyloid and its main constituent (beta-amyloid protein [Aβ]); immunostaining using antibodies specific to such proteins as ubiquitin, neuronal tau protein, and beta-amyloid protein; and visualization of β-pleated protein sheets using thioflavine S and Congo red with ultraviolet and polarized light. Tau (composed chemically of beta2-transferrin) is a discrete cytoskeletal protein that promotes the assembly of microtubules, stabilizes their structure, and participates in synaptic plasticity in a yet to be defined manner. In the pathologic circumstances of Alzheimer disease, progressive supranuclear palsy, and frontotemporal dementia (see further on), tau is hyperphosphorylated and aggregates, resulting in paired helical filaments that make up the neurofibrillary tangles. Electrophoretically, tau moves with the β2-globulins and is thought to function as a transferrin, that is it binds iron and delivers it to the cell. Its concentration can be measured in the CSF and serum, but this has not yet proven clearly to be useful as a diagnostic test.
The Aβ protein is a small portion of a larger entity, the amyloid precursor protein (APP), which is normally bound to neuronal membranes. As shown in Fig. 39-2, the Aβ protein is cleaved from APP by the action of proteases termed α, β, and γ secretase. One current hypothesis, developed by Selkoe and others, focuses on the manner in which APP is cleaved by these enzymes to give rise to different-length residues of Aβ. During normal cellular metabolism, APP is cleaved by either α or β secretase. The products of this reaction are then cleaved by the γ-secretase isoform of the enzyme. The sequential cleavage by α and then γ produces tiny fragments that are not toxic to neurons. However, cleavage by β and then γ results in a 40-amino-acid product, Aβ40, and a longer 42-amino-acid form. The latter Aβ42 form is toxic in several models of Alzheimer disease, and it has been proposed that the ratio of Aβ42 to Aβ40 is critical to the neuronal toxicity of amyloid.
Figure 39-2.
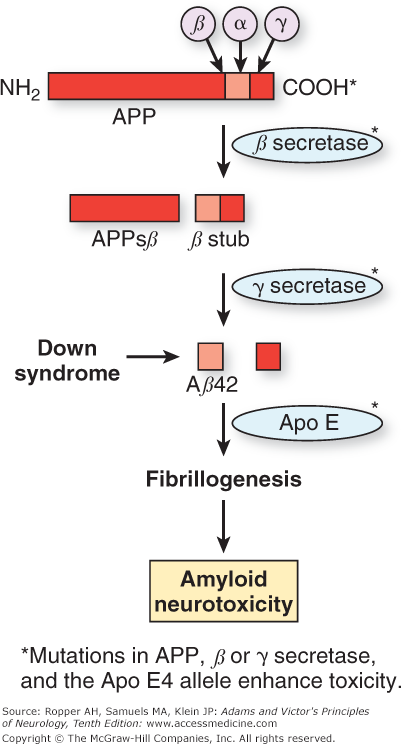
Diagram of proteolysis of amyloid precursor protein (APP). When APP is cleaved sequentially by β secretase and then secretase, the resulting amyloid protein can be 40 (Aβ40) or 42 (Aβ42) amino acids in length. The latter favors the formation of aggregated fibrillary amyloid protein (fibrillogenesis) rather than normal APP degradation. The fibrillary form of amyloid is neurotoxic, a mechanism favored as the cause of cell damage in Alzheimer disease. Formation of Aβ42 is promoted by mutations, either in the APP gene itself or in the presenilins. In Down syndrome, excess production of APP and its product Aβ42 is caused by triplication of the long arm of chromosome 21, the location of the APP gene. The Apo E4 allele is associated with inadequate clearance of Aβ42 and is another mechanism that promotes fibrillogenesis. (Modified by permission from Sisodia SS, St. George–Hyslop PH: γ-Secretase, notch, Aβ and Alzheimer’s disease: Where do the presenilins fit in? Nat Rev Neurosci 3:281–290, 2002.)
Several pieces of evidence favor the view that elevation of the levels of Aβ42 leads to aggregation of amyloid and then to neuronal toxicity. It appears that the diffuse deposition of Aβ42 precedes the formation of better-defined neurofibrils and plaques. The fact that the gene coding for APP is located on chromosome 21, one of the regions linked to one type of familial Alzheimer disease and the duplicated chromosome in Down syndrome, in which Alzheimer changes almost inevitably occur with aging (see further on), suggests that the overproduction of amyloid and all its Aβ residues are causative factors in the disease. Furthermore, the ratio of Aβ42 to Aβ40 is increased in Down syndrome. Another suggestive connection has been the finding that there are genetic defects in the genes encoding APP and in a pair of endosomal proteins termed presenilin 1 and 2 in some familial forms of Alzheimer disease. The presenilins interact with, or may be a component of, γ secretase, the enzyme that produces the Aβ42 fragment. Mutations of presenilin 1 and 2 also increase the relative levels of Aβ42. It should be noted that mutations of the APP and presenilin genes explain a very small proportion of Alzheimer cases (Terry). Transgenic mice that express human Alzheimer disease-associated mutations in APP or presenilin genes develop plaques with Aβ42 but not neurofibrillary tangles. Many of the relationships and mechanisms depicted in Fig. 39-2 are derived from the understanding of genetic forms of Alzheimer disease; the extent to which they will be implicated in the idiopathic disease is unknown. However, some form of disruption in these mechanisms is likely to be involved in the sporadic disease.
It must be emphasized, however, that there is still uncertainty regarding the relationship of amyloid deposition to the loss of neurons and brain atrophy. Alternatively, soluble oligomers of Aβ amyloid may be the toxic agents, whereas the emphasis until now has been on the effects of visible assemblies of insoluble amyloid fibrils. Similarly, TDP-43, the product of inadequate functioning of the progranulin gene, is also deposited in neurons and may play a substantial role in the severity of expression of Alzheimer disease; this protein has been implicated in the pathogenesis of frontotemporal dementia and motor neuron disease, both discussed later in the chapter. Others have questioned the amyloid hypothesis and pointed to the imprecise relationship between amyloid deposition and neuronal loss, even suggesting that aggregated amyloid is in some way a protective mechanism of cells.
The importance of neurofibrillary tangles has also been questioned, and the manner in which amyloid deposition relates to tangle formation is unclear. Unexplained also is prominent senile plaque formation in some cases and neurofibrillary tangles in others. One prevalent view is that the tangles are a secondary phenomenon. In their review, Hardy and Selkoe, authoritative investigators in this field, pointed out that “Although the amyloid hypothesis offers a broad framework to explain AD pathogenesis, it is currently lacking in detail, and certain observations do not fit easily with the simplest version of the hypothesis.” Nonetheless, the amyloid hypothesis is currently the strongest.
In recent years, some of the subcellular mechanisms that are deranged by the presence of intracellular or extracellular amyloid have been elucidated. The finding of a reduced number and enlargement of synapses in affected cortex early in the disease by DeKosky and Scheff and others could be interpreted as either the first sign of neuronal death or the result of the neuronal loss. Amyloid deposition would then be a later, secondary phenomenon. These are complex and uncertain connections but they are among the most promising findings in this field of research.
It was long ago established that Alzheimer disease is not caused by any of the usual types of arteriosclerosis. On the other hand, several studies have indicated that the presence of cerebral infarctions, small or large, and nondescript ischemic white matter disease accelerates the deposition of amyloid and the development of neurofibrillary tangles in the brains of Alzheimer patients (see further on); the mechanism of these interactions is not understood. Not surprisingly, cerebrovascular disease also exaggerates the rate of progression and degree of dementia. How this relates to the entity of arteriosclerotic, multiinfarct, or vascular dementia is entirely clear. Without doubt, as discussed in Chap. 34, multiple cerebral strokes cause increasing deficits that cumulatively qualify as a dementia. At least some of the focal lesions that contribute to the cognitive syndrome can be identified clinically and there is a stepwise decline in function that corresponds to strokes. Admittedly, this type of vascular dementia may be more difficult to recognize when a number of the infarcts are of the relatively silent lacunar type; the mental capacities of such patients may then appear to fail in a gradual and continuous fashion. Memory is relatively spared in the early stages and usually a pseudobulbar state or deterioration in gait accompanies the dementia. The subcortical white matter change of Binswanger disease causes similar diagnostic problems. We are inclined toward the view expressed in Chap. 21 and summarized in the commentary by Jagust that there is an undefined, and perhaps synergistic, interaction between strokes and progressive mental decline in patients with Alzheimer disease. Most often, in our experience, it is the degenerative condition of Alzheimer that explains the dementia. A similar relationship between Alzheimer disease and previous head injuries is tentative but has led to speculation that several types of brain injuries are conducive to the development of neurofibrillary tangles and amyloid deposition, perhaps as if they were part of a reparative response.
No relationship to premorbid personality traits earlier in life has been established, but an intriguing finding from what has become known as the “nun study” and several similar studies suggests that poorer linguistic capability early in life corresponded to the development of Alzheimer disease with aging (D.A. Snowden et al). In this study, the autobiographies of 93 nuns, written in their twenties, were rated for linguistic and ideational complexity. Of 14 sisters who died in late life, deterioration of cognitive function and neuropathologically proven Alzheimer disease occurred in 7 who had a low “idea density” in their writings and in none of 7 whose writings were cognitively more complex. Obviously this type of correlation is subject to several interpretations, but the general notion of “cognitive reserve” having either a protective property or simply hiding mental decline, has emerged from numerous other studies. Also, there has been a general perception confirmed by a few studies such as the one by Verghese and colleagues, that an active mental life may reduce the severity of mental decline with aging, but firm conclusions cannot be made from the available information.
Considerable interest was created in the late 1970s by the finding of a marked reduction in choline acetyltransferase (ChAT) and acetylcholine in the hippocampus and neocortex of patients with Alzheimer disease. This loss of cholinergic synthetic capacity was attributed to a reduction in the number of cells in the basal forebrain nuclei (mainly the nucleus basalis of Meynert), from which the major portion of neocortical cholinergic terminals originate (Whitehouse et al). However, a 50 percent reduction in ChAT activity has been found in regions such as the caudate nucleus, which shows neither plaques nor tangles (see review by Selkoe). The specificity of the nucleus basalis cholinergic changes has been questioned for other reasons as well. For one, Alzheimer brain also shows a loss of monoaminergic neurons and a diminution of noradrenergic, gabanergic, and serotonergic functions in the affected neocortex. The concentration of amino acid transmitters, particularly of glutamate, is also reduced in cortical and subcortical areas (Sasaki et al) and the concentration of several neuropeptide transmitters—notably substance P, somatostatin, and cholecystokinin are likewise low—but it has not been determined whether any of these biochemical abnormalities, including the cholinergic ones, are primary or secondary to heterogeneous neuronal loss. Nevertheless, the administration of cholinomimetics—either acetylcholine precursors (e.g., choline or lecithin), degradation inhibitors (e.g., physostigmine), or muscarinic agonists that act directly on postsynaptic receptors—have had a mild and unsustained therapeutic effect (see further under “Treatment”).
Chase and associates have demonstrated a 30 percent reduction in cerebral glucose metabolism in Alzheimer disease, greatest in the parietal lobes, but this seems most likely to be secondary to tissue loss in these regions. Even if not of pathogenic significance, it finds value as a diagnostic marker of the disease. The role of aluminum in the genesis of neurofibrillary tangles, as was once proposed, has never been validated. It has been suggested that the use of estrogen by postmenopausal women or of antiinflammatory agents in men or women delayed the onset of the disease or reduced its occurrence, but neither of these have been corroborated by other studies.
Of great importance was the aforementioned series of discoveries in patients with inherited forms of Alzheimer disease, of defective genes that code for errant APPs localized to chromosome 21 near the β-amyloid gene (St. George-Hyslop et al). As mentioned, this also provided an explanation for the Alzheimer changes that characterize the brains of practically all patients with the trisomy 21 defect (Down syndrome) who survive beyond their twentieth year; they overproduce amyloid as a result of the triplication of the gene. But gene defects on chromosome 21 are responsible for only a small proportion of familial cases and a minuscule percentage of disease overall. Other kindreds with familial Alzheimer disease have been linked to rare dominant mutations of the presenilin genes on chromosome 14 (presenilin 1; Sherrington et al), accounting in some series for up to 50 percent of familial cases, and on chromosome 1 (presenilin 2), which may account for many of the remaining ones (Levy-Lahad et al). These are summarized in Table 39-1. The age of onset of the disease in these familial forms, as in the Down cases, is earlier than that in sporadic forms. These cohorts of patients have provided great insight into long duration between the appearance of amyloid in the brain, approximately a decade, and the onset of clinical disease, and they suggest the potential use of imaging of chemical biomarkers for the disease (The Dominantly Inherited Alzheimer Network; see Bateman et al).
GENE | PROTEIN | INHERITANCE | AGE | CLINICAL FEATURES |
|---|---|---|---|---|
APP | Amyloid precursor protein | AD | Early | Rare but clinically simulates sporadic Alzheimer disease |
PS1 | Presenilin 1 | AD | Early | As above |
PS2 | Presenilin 2 | AD | Early | As above |
Apo E | Apolipoprotein E | Haplotype | Late | Modifies susceptibility to Alzheimer disease; ϵ-4 allele represents risk |
UBQLN1 | Ubiquilin 1 | SNP | Late | Familial cases only |
TREM2 | TREM2 | SNP | Late | Modulating factor as for Apo E |
It has been clear for some time that an excess or aberrant amyloid alone is an incomplete explanation for the disease. Certain sequence variants in normal genes confer an increased risk of the disease. The one first discovered was Apo E, a regulator of lipid metabolism that has an affinity for Aβ in Alzheimer plaques, has been found to modify the risk of acquiring Alzheimer disease. Of the several isoforms of Apo E, the presence of E4 (and its corresponding allele e4 on chromosome 19) is associated with a tripling of the risk of developing sporadic Alzheimer disease (Roses; Strittmatter et al; Polvikoski et al). This is the same allele that contributes to an elevated low-density lipoprotein fraction in the serum. Possession of two e4 alleles virtually assures the development of disease in those who survive to their eighties. The e4 allele also modifies the age of onset of some of the familial forms of the disease. In contrast, the e2 allele is underrepresented among Alzheimer patients. For these reasons it has been proposed that Apo E, by interacting with APP or tau protein in some way, modifies the formation of plaques. Indeed, possession of the e4 allele correlates with increased deposition of Aβ in the brain (McNamara). As pointed out by Hardy, Apo E appears to act at a point in the pathogenesis that is after the various genetic mutations have influenced the cellular pathology that ostensibly causes Alzheimer disease. However, these statistical relationships do not invariably connect an allele to the disease in a particular individual. In other words, the e4 allele does not act as a mendelian trait but as a susceptibility (risk) factor. It follows that many, if not most, individuals who develop Alzheimer disease do not have the risk allele. Moreover, many individuals with the e4 allele live into their seventies and eighties without developing Alzheimer disease. All that can be stated with certainty is that, on average, the presence of the e4 allele accelerates the appearance of Alzheimer disease by about 5 years.
Another polymorphism in TREM2 is quite rare in comparison to the aforementioned Apo E variants but confers an equivalent risk of Alzheimer disease that has been shown in several populations in (Guerreiro et al and Jonsson et al). In sporadic Alzheimer disease, the TREM2 polymorphism that is implicated in Alzheimer disease putatively causes inadequate phagocytic clearance of amyloid. Another rare modifying gene has been found in familial cases at the UBQLN1 (ubiquilin 1) site, coding for a protein that interacts with PS1 and PS2 and participates in proteasomal degradation.
Studies with CT and MRI are useful, but not definitive ancillary tests (Fig. 39-3). In patients with advanced Alzheimer disease, the lateral and third ventricles are enlarged to about twice the normal size and the cerebral sulci are proportionately widened. Coronal MRI of the medial temporal lobes may reveal a disproportionate atrophy of the hippocampi and a corresponding enlargement of the temporal horns of the lateral ventricles. Early in the disease, however, the changes do not exceed those found in many mentally intact old persons. For this reason, one cannot rely solely on imaging procedures for diagnosis and CT and MRI are most valuable in excluding alternative causes of dementia such as brain tumor, subdural hematoma, cerebral infarction, and hydrocephalus. The EEG undergoes mild diffuse slowing, but only late in the course of the illness; it is useful again, in the exclusion of alternative causes of mental decline that manifest themselves in seizure activity or changes typical of metabolic encephalopathy. The CSF is also normal, although occasionally the total protein is slightly elevated. Using the constellation of clinical data, cerebral imaging in the context of the age of the patient and time course of the disease, the diagnosis of dementia of Alzheimer type is made correctly in 85 to 90 percent of cases.
Figure 39-3.
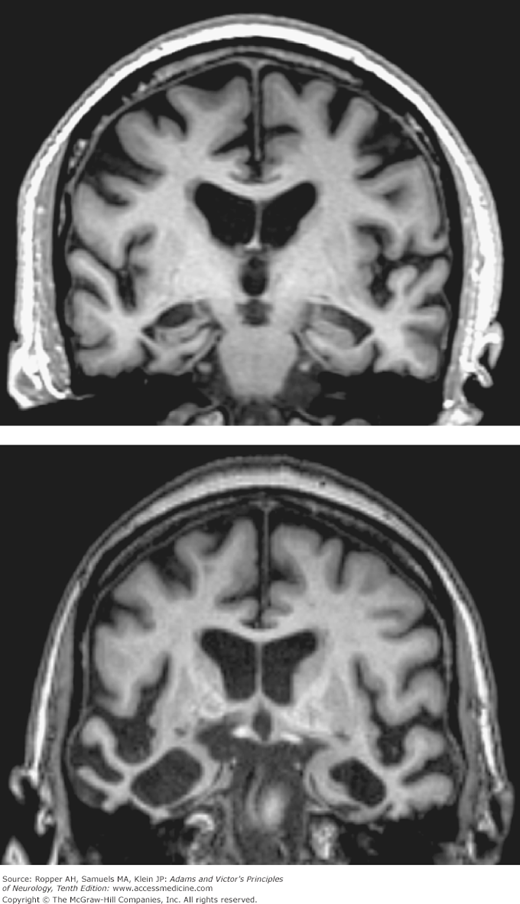
Top: Coronal T1-weighted MRI of a 74-year-old man with moderate Alzheimer-type dementia. Diffuse cerebral and hippocampal atrophy with ex vacuo ventricular and cortical sulcal dilation is noted. Bottom: Coronal T1-weighted MRI of a 70-year-old woman with behavioral variant frontotemporal lobar dementia. Atrophy of the right greater than left temporal lobes is out of proportion to atrophy of the frontal and parietal lobes.
Of considerable value have been studies of cerebral blood flow single-photon emission computed tomography [SPECT]) and metabolism (positron emission tomography [PET]), which early in the illness often, but not always, show diminished activity in the parietal association regions and the medial temporal lobes. In most cases, when such changes are evident, the diagnosis was already obvious on clinical grounds. Newer PET ligand agents that bind to amyloid, such as the “Pittsburgh compound” and tau-ligands are more sensitive in identifying and observing the course of Alzheimer disease. Their main utility may be in detecting changes before brain atrophy is evident and in identifying patients who have the earliest changes of Alzheimer disease, whose disease course may be amenable to alteration by medications.
Neuropsychologic tests in the typical case show disproportionate deterioration in memory and verbal access skills. Testing is particularly useful when there is a serial decline in ability. Certain aspects of attention and executive function in Alzheimer disease that also show changes in Alzheimer disease were reviewed by Perry and Hodges. The use of these examinations is described in Chap. 21.
There are no established biologic markers of Alzheimer disease with the possible exception of the ratio of Aβ 42 to tau, in the cerebrospinal fluid (the ratio is low in Alzheimer disease). This test is used in some clinics, but may not be well enough validated for routine use (Maddalena et al). Schoonenboom and colleagues have shown that the incorporation of CSF phosphorylated tau (p-tau) with the typical CSF amyloid/tau ratio may provide additional specificity in distinguishing Alzheimer from other dementing diseases.
Formerly, when virtually all forms of dementia were untreatable, there was little advantage to either the patient or the family in ascertaining the cause of the cerebral disease. There are now adequate treatments for a number of diseases and conditions that cause cognitive decline, putting a premium on proper diagnosis.
The currently potentially treatable forms of dementia are those caused by normal-pressure hydrocephalus; chronic subdural hematoma; the dementia of AIDS; paraneoplastic and related autoimmune encephalitis; nutritional deficiencies (thiamine—Wernicke-Korsakoff syndrome, Marchiafava-Bignami disease, pellagra, vitamin B12 deficiency); chronic intoxication (e.g., alcohol, sedatives); multiple cerebral infarctions; certain endocrine and metabolic disorders (myxedema, Hashimoto encephalopathy), neurosyphilis and other chronic meningitides, Cushing disease, chronic hepatic encephalopathy; frontal and temporal lobe tumors; vascular dementia, cerebral vasculitis; sarcoidosis; progressive multifocal leukoencephalopathy (PML), Whipple disease; multiple sclerosis; and sometimes neglected, the pseudodementia of depression. Exclusion of most of these diseases is readily accomplished by careful history, sequential clinical evaluations, and testing of blood and CSF, EEG, CT, MRI, and neuropsychologic testing can be undertaken. We have regularly but infrequently incorporated the results of metabolic brain imaging (both FDG-PET and amyloid-ligand imaging) as well as CSF amyloid-tau ratio. We anticipate that these tests or similar ones may find more frequent use. In exceptional situations, brain biopsy may be justified in the diagnosis of dementia, almost limited to rapidly progressive cases. A perspective, albeit from a sample that cannot be generalized to practice, has been given by Warren and colleagues of 90 consecutive brain biopsies performed between 1989 and 2003 for the evaluation of dementia. More than half provided a diagnosis, mostly Alzheimer, Creutzfeldt-Jakob disease, and inflammatory disorders. However, reasonable assurances must be given to the neurosurgeon that prion disease is unlikely.
One problem in differential diagnosis is the distinction between a late-life depression and a dementia, especially when some degree of both is present. Observation over several weeks or more, and the patient’s demeanor, makes the distinction clearer. Multiinfarct dementia is usually not difficult to separate from Alzheimer dementia, as discussed further on. The dementia of normal-pressure hydrocephalus may also be confused with Alzheimer dementia (see Chap. 30). The problem of distinguishing Alzheimer disease from a more “benign” form of memory decline associated with aging comes up frequently in practice, as discussed further on. These treatable conditions are discussed in Chaps. 21, 30, and 34 and the important topic of depression is addressed in Chap. 52. Often we have been confident on clinical grounds that a patient had Alzheimer disease, only to have revealed at autopsy that progressive supranuclear palsy, Lewy-body disease, Pick disease, another non-Alzheimer degeneration of the frontal lobes, or cortical-basal-ganglionic degeneration was the cause. All are discussed later in this chapter.
There is no evidence that any of the formerly proposed therapies for Alzheimer disease—cerebral vasodilators, stimulants, L-dopa, massive doses of vitamins B, C, and E, gingko biloba, hyperbaric oxygen, intravenous immunoglobulin, and many others—have any salutary effect. Trials of oral physostigmine, choline, and lecithin have yielded mostly negative or uninterpretable results.
The effect of the currently used cholinergic precursors and agonists and acetylcholinesterase inhibitors, such as donepezil, is modest. With regard to the latter group of drugs, several large trials have demonstrated a slight prolongation of the patient’s ability to sustain an independent life, but such evidence generally requires that the medications be taken for 6 to 12 months. For example, a meta-analysis of the drugs collectively demonstrated a mean improvement of 2 to 3 points on the 70-point Alzheimer Disease Assessment Scale and a slight delay in progression. Despite some trials that have failed to demonstrate benefit (c.f., AD 2000 Collaborative Group), the balance of evidence favors the use of these medications in practice, but only in mildly or moderately affected patients.
Side effects of the aforementioned class of drugs may include nausea and less often, vomiting. The families of our patients report from time to time that the medication caused insomnia or increased confusion. It is worth mentioning that when the acetylcholine receptor antagonist succinylcholine is used prior to general anesthesia, its effects may be prolonged in patients taking the above drugs. The use of trazodone, haloperidol, thioridazine, risperidone, and related drugs may suppress some of the aberrant behavior and hallucinations when these are problems, making life more comfortable for both patient and family, but several trials suggest that their general application causes more problems than it solves and they must often be discontinued in response to adverse effects. The randomized trial conducted by Schneider and coworkers found that olanzapine, quetiapine, and risperidone for the treatment of psychosis, aggression, or agitation with Alzheimer disease were approximately as good as placebo in relieving these symptoms, but largely because the drugs were not tolerated. Olanzapine was slightly preferable in those who continued taking the medication. The clinician is left with little recourse but to use this class of medications or haloperidol to control unmanageable behavior. Small doses of diazepines, such as lorazepam, are useful when sleep is severely disturbed, but they often increase confusion as well.
The N-methyl-D-aspartate (NMDA) glutaminergic antagonists, specifically memantine (20 mg daily), have also been tried. In a study of memantine by Reisberg and colleagues of 252 patients (187 of whom completed the trial), there were better results on a few scales that reflected functional behavior compared to the use of placebo, but there was no change in 3 main measures of cognitive performance. Because the side effects were ostensibly minor, this drug has been approved for use in late-stage Alzheimer disease and in conjunction with cholinergic drugs. Nevertheless, hallucinations or agitation may occur and require discontinuation. The combination of memantine and donepezil in moderately to severely affected patients offered no benefit over either drug alone (Howard et al). The effects of these drugs in later stages of the disease are, in any case, minimal.
A provocative series of studies using a small molecule inhibitors of the enzyme γ-secretase (semagacestat; see Doody et al, 2013), and a monoclonal antibody directed at soluble forms of amyloid (solanezumab; see Doody et al, 2014) have failed to demonstrate clear benefit in early Alzheimer disease. The presumption is that such agents might be useful if started in the presymptomatic stages of disease.
A series of animal experiments that demonstrated the possibility of removal of plaques by immunization against amyloid has led to human studies with a similar vaccination. One trial was stopped because of the occurrence of an immune encephalitis in a small number of patients, but in autopsy material there were indications that this novel approach may have had the desired effect of reducing amyloid deposition (Orgogozo et al). Revised vaccines are being formulated for further testing of this approach.
Given the state of therapeutics for Alzheimer disease, always important is the general management of the demented patient, which should proceed along the lines outlined in Chap. 21, keeping in mind that the physician’s counsel is often the family’s main resource for important medical and social decisions.
As indicated earlier, the histologic changes of Alzheimer disease have a number of interesting associations. Amyloid plaques and tangle deposition are far more common in the brains of patients with Parkinson disease (20 to 30 percent) than in the brains of age-matched controls (Hakim and Mathieson). These findings partly explain the high incidence of dementia in patients with Parkinson disease (see further on). As mentioned, with the advance of Alzheimer disease, extrapyramidal features may emerge. In such cases, Burns and colleagues have found changes in the substantia nigra including accumulation of synuclein and tau representative of Lewy bodies. Another association between the 2 diseases is apparent in the Guamanian Parkinson–dementia complex, which is also discussed below. In this entity, the symptoms of dementia and parkinsonism are related to neurofibrillary changes in the cerebral cortex and substantia nigra, respectively; senile plaques and Lewy bodies are unusual findings. What can be deduced from the crossover syndromes is that multiple degenerative changes can occur in these diseases and give rise to heterogeneity in clinical presentation.
The finding of neurofibrillary tangles (and to a lesser extent of plaques) in boxers (“punch-drunk” syndrome, or dementia pugilistica) is another interesting ramification of the Alzheimer disease process in that trauma appears to be able to elicit one of the core features of the disease as discussed in Chap. 35. Some cases of primary progressive aphasia (see further on) have Alzheimer change and amyloid plaque deposition as the primary pathologic change. There are other unusual and meaningful associations, such as dementia with motor neuron disease or the cases of familial dementia with spastic paraplegia reported by Worster-Drought and by van Bogaert and their associates (see later in this chapter). Here, neurofibrillary change is the most prominent feature whereas amyloid plaques are negligible in number or absent.
Another provocative connection is the already mentioned interrelationship between cerebrovascular disease and Alzheimer disease. This is a complex area that at one time, considered the 2 processes to be intimately related and later, was rejected, only to now be resurrected with clearer focus, as discussed Chap 34.
This broad category of disease has evolved and the nosology is confusing because type of selective atrophy of a cerebral lobe may be caused by several different histopathologic changes. The notion of lobar atrophy was introduced in 1892 when Arnold Pick of Prague described a special form of cerebral degeneration in which the atrophy was circumscribed (most often in the frontal or temporal lobes), with involvement of both gray and white matter; hence the term he applied was lobar rather than cortical sclerosis. In 1911, Alzheimer presented the first careful study of the microscopic changes, followed by even more complete analyses of the pathologic changes by the prominent neuropathologists of the age. As mentioned the pathologic change associated may be any one of several types: Pick inclusion bodies, neurofibrillary tangles, other inclusions, or with no characteristic changes except for neuronal loss. Contrariwise, gliosis and mild spongiform changes in the superficial layers of cortex, and even typical plaque and tangle pathology, have all been associated with syndromes of gross atrophy of the frontal or temporal lobes. What has emerged since his work is that the most common and important of the lobar atrophies is a group of frontotemporal degenerations that have diverse clinical and pathologic profiles.
In contrast to Alzheimer disease, in which the atrophy is relatively diffuse, the pathologic change in lobar atrophy is circumscribed and often asymmetrical. The parietal lobes are involved less frequently than the frontal and temporal lobes. The affected gyri become paper thin, resembling, in the advanced stages, the kernel of a dried walnut. The cut surface reveals not only a marked narrowing of the cortical ribbon but a grayish appearance and reduced volume of the underlying white matter. The corpus callosum and anterior commissure share in the atrophy but almost certainly as secondary phenomena. The overlying pia-arachnoid is often thickened, and the ventricles are enlarged. The pre- and postcentral, superior temporal, and occipital convolutions are relatively unaffected and stand out in striking contrast to the wasted parts.
The more common frontotemporal lobar degenerations (FTLDs) (to which Pick’s name was attached) may display any one of several pathologic changes and reflect different genetic causes. For example, the behavioral or the aphasic variants of FTD, which are described below, may be the result of the deposition of tau, progranulin, amyloid, or synuclein. It is not clear to us if the term “Pick disease” is worth retaining to denote a unique process aside from the unusual type that is due to deposition of argyrophilic intracytoplasmic inclusions (Pick bodies) and diffusely staining ballooned neurons (Pick cells). In other respects, it is simply one of the large group of FTLDs. It is the lobar atrophy and marked changes in the underlying white matter that provide the unifying elements of this group of diseases.
The descriptive terms frontotemporal lobar atrophy and frontotemporal dementia are used by neurologists and neuropathologists to refer to a clinical syndrome that is associated with degeneration of the frontal and temporal lobes. Some of the clinical aspects of frontotemporal dementia were discussed in Chap. 21, but broadly speaking, there are 2 main types: a behavioral variant and a language variant, the latter being divided into semantic dementia, progressive nonfluent aphasia, and a logopenic variant, all described below.
Patients under consideration with behavioral changes present with personality and related abnormalities that include apathy, disinhibition, perseveration, poor judgment and limited ability for abstraction, loss of empathy, bizarre affect, eating disorders, and a general disengagement. Insight is almost always impaired and some subjects become euphoric or display repetitive compulsive behaviors. An initial diagnosis of depression has been common. Other psychiatric symptoms such as sociopathic and disinhibited behavior with aspects of hyperorality and hyperphagia may predominate late in the illness. Utilization behavior (the compulsive use of implements and tools put before the patient) is also displayed in advanced cases.
CT, MRI, and functional imaging demonstrate a disproportionate atrophy and hypofunction in the frontal lobes, usually asymmetric. A proportion of patients with this type of frontotemporal dementia have parkinsonian features. A form of motor neuron disease is also linked to frontotemporal dementia in a small number of cases. This is particularly the case in the Guamanian (now called western Pacific) variety and in the heredofamilial frontotemporal atrophy linked to a mutation on chromosome 17.
In some writings on this subject, the term frontotemporal dementia has come to be used in a highly restricted sense, being assigned to cases that show only tau-staining material in neurons. Most of the cases are sporadic, but the inherited variety linked to chromosome 17, in which parkinsonism is prominent, supports its distinction as a separate entity; it is in these cases that the intraneural deposition of tau is most striking, in both the frontotemporal cortex and the substantia nigra. In a few familial cases, this process is attributable to mutations in the gene on chromosome 17 that encodes the tau protein. These mutations alter the proportions of different isoforms of this protein and lead both to tau accumulation and its hyperphosphorylation. Indeed, many cases of frontotemporal dementia are associated with tau gene mutations. However, abnormal aggregates of tau have been identified in practically all neurodegenerative atrophies and, of course, form the main constituent of the paired helical filaments (neurofibrillary tangles) of Alzheimer disease, and in progressive supranuclear palsy where they are abundant, although of slightly different structure. From the observations of Brun and Passant and of Neary and associates, pure tau-reactive cases outnumber Pick disease when the latter is strictly defined by the cortical white matter degeneration and Pick inclusions.
Nonetheless, a frontotemporal dementia identical to that of the tau-reactive cases has been observed in others without any tau or synuclein staining of neurons. Many of the frontally predominant cases have shown deposition of the protein progranulin, consisting mainly of a ubiquitin neuronal inclusion consisting of TDP-43 (TAR DNA-binding protein), the result of PGRN mutations.
Focal disturbances, particularly aphasia and apraxia, occur early and prominently in certain patients with lobar degenerations, indicating a lesion in the left frontal or temporal lobes. Viewed from another perspective, a prominent language disorder has been described in almost two-thirds of all patients with temporal lobe atrophy.
Several types of this disturbance have been delineated. In the first, progressive nonfluent aphasia, the patient initially speaks less and has word-finding difficulty (anomia), but language structure is intact (Mesulam, 1982); later, he may forget and misuse words and soon fails to understand much of what is heard or read. Sentences are short and telegraphed. Later, dysarthria and apraxia become apparent and finally, the patient is virtually mute, seemingly without impulse to speak, and with an inability to form words (Snowden et al, 1992).
A second type, semantic dementia, is characterized by early difficulty naming items, people, and words, followed by verbal perseveration, but fluency is retained. There is considerable difficulty in generating lists of words of a given category, such as animals. These individuals are quite aware that they are having trouble finding words. Eventually the patient loses not just the use of names of people and objects, but also their meaning, or the conceptual knowledge of the word. Some may develop severe prosopagnosia, especially if the atrophy is predominantly right sided. Memory for day-to-day events is preserved.
A third type has been proposed, logopenic aphasia, that shares most aspects of nonfluent aphasia but in which the meaning of words is retained.
According to Mesulam (2003), who has studied the condition extensively, 60 percent of these cases show no characteristic pathologic change, 20 percent have Pick bodies, and a similar proportion show the typical changes of Alzheimer disease in the affected cortical region. A clear familial tendency has not been found. Chapter 23 can be consulted for details of the aphasic disorders.
This regional variant of lobar degeneration has been slightly less frequent than primary progressive aphasia in our practices. The fundamental feature is the progressive loss of the ability to understand and use visual information. The result is progressive and ultimately severe visuospatial difficulty with a relative preservation of memory. Prosopagnosia, achromatopsia, and dyslexia emerge, or, there may be difficulty with depth perception, reaching for objects and an inordinate sensitivity to bright light. Patients under our care have initially had a vague sense of visual disorientation followed over months by difficulty in seeing or recognizing objects in front of them. Many have alexia with agraphia while others have acalculia or the other elements of the Gerstmann syndrome. Several eventually become cortically blind. The syndrome is essentially that of an apperceptive visual disturbance that includes fragments of the Balint and the Gerstmann syndromes. The average age of onset is about 60 years. The most common pathologic change in most reports has been characteristic of Alzheimer disease.
Next to Alzheimer disease, diffuse Lewy-body disease, or Lewy-body dementia, has been the most frequent pathologic diagnosis established in many series of globally demented patients. Reports of this condition have been increasing steadily since the original communication by Okazaki and colleagues in 1961 (see review by Kosaka). The disease is defined by the diffuse involvement of cortical neurons with Lewy-body inclusions and by an absence or inconspicuous number of neurofibrillary tangles and amyloid plaques. To some extent, increased recognition of this disorder is a result of improved histologic techniques, particularly the ability to detect ubiquitin and synuclein, main components of the Lewy body, by immunostaining. With this improved detection has come a better definition of the clinical syndrome and its distinctions from Alzheimer and other dementias. Because the Lewy bodies in cortical neurons are not surrounded by a distinct halo, as they are in the substantia nigra in cases of Parkinson disease (see further on for discussion and photomicrograph of a typical Lewy body) they were not readily appreciated. Aggreagated α-synuclein is the main component of the Lewy body an observation that will prove important in understanding both Parkinson disease and Lewy-body dementia.
The disease in its typical form is marked by parkinsonian features, dementia, and a tendency to episodic delirium, especially nocturnally, and rapid eye movement (REM) sleep behavior disorder (described below and in Chap. 19). Diagnostic criteria have been offered by a working group, requiring 2 of 3 of the following: a parkinsonian syndrome (usually symmetric), fluctuations in behavior and cognition, and recurrent hallucinations (McKeith et al). The latest recursion of this group’s criteria emphasizes the presence of the REM sleep behavior disorder and severe neuroleptic sensitivity.
In an analysis of 34 cases of diffuse Lewy-body disease, Burkhardt and colleagues, found that the most characteristic syndrome was one of progressive dementia in an elderly patient with the additional late onset of parkinsonism in many cases. In Lennox’s summary of 75 cases, parkinsonism, particularly with limb and axial rigidity, was a prominent feature in 90 percent once the illness was fully developed, and almost half had tremor of the parkinsonian type (this is somewhat different from other series). Byrne and associates, as have many others, pointed out that episodic confusion, hallucinations, and paranoid delusions were features of Lewy-body dementia; such psychotic aspects are generally uncharacteristic of Alzheimer and lobar dementias, and only then, in advanced stages. In Lennox’s review, one-third of patients had these swings in behavior, but as the illness advanced, amnesia, dyscalculia, visuospatial disorientation, aphasia, and apraxia differed little from those of Alzheimer disease. In the cases reported by Fearnley and coworkers, there was a supranuclear gaze palsy simulating that of progressive supranuclear palsy. These overlapping clinical features make diagnosis difficult unless the specific feature of episodic hallucinations is evident. Difficulty in diagnosis also arises because the parkinsonian disorder may be either mild or prominent and may occur as an early or a late manifestation.
The parkinsonian features can respond favorably to L-dopa, but only for a limited time and sometimes at the expense of causing an agitated delirium or hallucinations that would be uncharacteristic of early Parkinson disease (Hely et al); in others, the response to L-dopa is inconsistent or inapparent. Some patients also have orthostatic hypotension corresponding to cell loss and Lewy bodies in the intermediolateral cell column of the spinal cord or in the sympathetic ganglia, thereby simulating striatonigral degeneration or Shy-Drager syndrome (see further on). Others have commented on an extreme sensitivity of such patients to neuroleptic drugs, including increased confusion and greatly worsening parkinsonism or the development of the neuroleptic malignant syndrome.
In our experience with Lewy-body disease, the parkinsonian symptoms have been more prominent than they are in progressive supranuclear palsy, and the most characteristic feature besides the movement disorder and a slowly advancing dementia has been a vacuous, anxious state with intermittent psychotic or delirious behavior.
At least one randomized trial has described benefit from the anticholinesterase inhibitor, rivastigmine, in reducing delusions, hallucinations, and anxiety (McKeith and colleagues, 2000). With regard to diagnostic testing, the finding of reduced activity in the posterior parietal cortical regions on PET scans (as in Alzheimer disease) has been found as a relatively consistent, but not invariable, feature.
This obscure entity has been connected with a late-life dementia in which behavioral disturbances precede memory difficulty. Whether the finding of argyrophilic grains in the mediotemporal lobe, different from tau-laden neurofibrillary tangles and from the glial inclusions (putatively a defining feature of multiple system atrophy), constitutes a specific entity is not clear to the authors. The finding overlaps with the deposition of other materials that are more closely associated with dementing diseases such as phosphorylated tau and Lewy bodies. Probst and Tolnay remarked that these small argyrophilic inclusions are not found in nondemented individuals. It is unlikely that the condition can be identified in life; if it is a genuine entity, it must be rare. The interested reader may consult the review by Ferrer.
There have been infrequent case reports of dominantly inherited, adult-onset dementia with a fulminant evolution suggestive of encephalopathy and the special feature of seizures. The distinctive feature has been the presence at autopsy of large eosinophilic, PAS-positive intraneuronal inclusions that contain aggregates of neuroserpin, thus the initial description of “familial encephalopathy with neuronal inclusion bodies.” The serpins are a family of protease inhibitors that include neuroserpin, a protein expressed exclusively in neurons, and α1-antitrypsin. The neuronal inclusions are densest in the deep layers of the cortex and in the substantia nigra. Missense mutations in the gene encoding neuroserpin have been identified as the cause. This entity is reviewed by Lomas and Carrell.
Dementing Diseases in Which Other Neurologic Abnormalities Are Prominent
This disease, distinguished by the triad of dominant inheritance, choreoathetosis, and dementia, commemorates the name of George Huntington, a medical practitioner of Pomeroy, Ohio. In 1872, his paper, read before the Meigs and Mason Academy of Medicine and published later that year in the Medical and Surgical Reporter of Philadelphia, gave a succinct and graphic account of the disease that was based on observations of patients that his father and grandfather had made in the course of their practice in East Hampton, Long Island. Reports of this disease had appeared previously (see DeJong for historical background) but they lacked the completeness of Huntington’s description. In 1932, Vessie was able to show that practically all the patients with this disease in the eastern United States could be traced to about 6 individuals who had emigrated in 1630 from the tiny East Anglian village of Bures, in Suffolk, England. One remarkable family was traced for 300 years through 12 generations, in each of which the disease had expressed itself.
To quote Huntington, the rule has been that “When either or both of the parents have shown manifestations of the disease, one or more of the offspring invariably suffer of the disease, if they live to adult life. But if by any chance these children go through life without it, the thread is broken and the grandchildren and great grandchildren of the original shakers may rest assured that they are free from disease.” Davenport, in a review of 962 patients with Huntington chorea, found only 5 who had descended from unaffected parents. Possibly, in these 5 patients, a parent had the trait, in very mild form, or parentage was in question, because spontaneous mutations are rare.
In university hospital centers, this is a regularly observed type of hereditary nervous system diseases and the main cause of progressive chorea at most ages. Its overall frequency is estimated at 4 to 5 per million, and 30 to 70 per million among whites of northern European ancestry. The usual age of onset is in the fourth and fifth decades, but 3 to 5 percent begin before the fifteenth year and some even in childhood, where it takes on special form. In approximately 30 percent, symptoms become apparent after 50 years. The progression of the disease is generally slower in older patients for reasons noted below. Once begun, the disease progresses relentlessly, until only a restricted existence is possible and a medical disease terminates life.
Exhaustive genealogic documentation many years ago established the cause to be an autosomal dominant gene with complete penetrance. Koller and Davenport made the observation that young patients usually inherit the disease from their fathers and older patients from their mothers. It has been observed beginning at almost the same age in identical twins.
The first important achievement in respect to the biologic understanding of Huntington disease was the discovery by Gusella and colleagues of a marker linked to the Huntington gene and localized to the short arm of chromosome 4. Subsequently, these investigators and others identified the mutation as an excessively long repeat of the trinucleotide CAG within the Huntington gene, the length (number) of which determines not only the presence of the disease, but also the age of onset, longer repeat lengths being associated with an earlier appearance of signs. At the Huntington gene locus there are normally 11 to 34 (median: 19) consecutive repetitions of the CAG triplet, each coding for glutamine. Individuals with 35 to 39 triplets may eventually manifest the disease but it tends to be late in onset and mild in degree, or limited to the below-mentioned “senile chorea.” Those with more than 42 repeats almost invariably acquire the signs of disease if they live long enough. The rare alternative mutation, termed HDL2 (Huntington disease-like-2), is associated with CATCG repeat expansion of the juntophilin-3 gene, but it is so infrequent that few clinicians will encounter it (Margolis et al).
These discoveries have made possible the development of a genetic test for the measurement of the repeat length that confirms the diagnosis in symptomatic patients and allows screening of asymptomatic individuals. Because there is no treatment for the disease, testing raises certain ethical considerations that must be resolved before its widespread utilization.
The mental disorder assumes several subtle forms long before the more obvious deterioration of cognitive functions becomes evident. In approximately half the cases, slight but annoying alterations of personality are the first to appear. Patients begin to find fault with everything, to complain constantly, and to nag other members of the family; they may be suspicious, irritable, impulsive, eccentric, untidy, or excessively religious, or they may exhibit a false sense of superiority. Poor self-control may be reflected in outbursts of temper, fits of despondency, slovenliness, alcoholism, or sexual promiscuity. Disturbances of mood, particularly depression, are common (almost half of the patients in some series) and may constitute the most prominent symptoms early in the disease. Invariably, sooner or later, the intellect begins to fail globally. The patient becomes less communicative and socially withdrawn. The emotional disturbances and changes in personality may reach such proportions as to constitute a virtual psychosis with persecutory delusions or hallucinations.
Diminished work performance, inability to manage household responsibilities, and disturbances of sleep may prompt medical consultation. There is difficulty in maintaining attention and concentration and in assimilating new material. Mental flexibility lessens. Simultaneously, there is loss of fine manual skills (see further on). The performance parts of the Wechsler Adult Intelligence Scale show greater loss than the verbal parts. Memory is relatively spared. This gradual dilapidation of intellectual function has been characterized as a “subcortical dementia,” that is elements of aphasia, agnosia, and apraxia are observed only rarely and memory loss is not profound. Often the process is so slow, particularly in cases of late onset, that a fair degree of intellectual capacity seems to be retained for many years.
The abnormality of movement is subtle at first and most evident in the hands and face; often the patient is merely considered to be fidgety, restless, or “nervous.” Slowness of movement of the fingers and hands, a reduced rate of finger tapping, and difficulty in performing a sequence of hand movements are early signs. Gradually these abnormalities become more pronounced until the entire musculature is implicated with chorea. The frequency of blinking is increased (the opposite of parkinsonism), and voluntary protrusion of the tongue, like other attempts at sustained posture, is constantly interrupted by unwanted darting movements. In the advanced stage of the disease the patient is seldom still for more than a few seconds. The choreic movements are slower than the brusque jerks and postural lapses of Sydenham chorea, and they involve many more muscles. They tend to recur in stereotyped patterns yet are not as stereotyped as tics. In advanced cases, they acquire an athetoid or dystonic quality. Muscle tone is usually decreased until late in the illness, when there may also be some degree of rigidity, tremor, and bradykinesia, elements suggestive of Parkinson disease. Parkinsonism with rigidity characterizes the Westphal or “rigid” variant, which is more common with a childhood onset, or the HDL2 genetic variant noted earlier. Tendon reflexes are exaggerated in one-third of patients, but only a few have Babinski signs. Voluntary movements are initiated and executed more slowly than normal, but there is no weakness and no ataxia, although speech, which becomes dysarthric and explosive because of incoordination between tongue and diaphragm, may convey the impression of a cerebellar disorder. Inability to hold the tongue protruded is characteristic. In late-onset cases there may be an almost constant rapid movement of the tongue and mouth, simulating the tardive dyskinesia that follows the use of neuroleptic drugs. These disorders of movement that characterize Huntington chorea are described more fully in Chap. 4.
Oculomotor function is subtly affected in most patients (Leigh et al; Lasker et al). Particularly characteristic are impaired initiation and slowness of both pursuit and volitional saccadic movements and an inability to make a volitional saccade without movement of the head. Excessive distractibility may be noticed during attempted ocular fixation. The patient feels compelled to glance at extraneous stimuli even when specifically instructed to ignore them. Upward gaze is often impaired as the illness progresses.
As Wilson stated, the relation of the choreic to the mental symptoms “abides by no general rule.” Most often the mental symptoms precede the chorea but they may accompany or follow it, sometimes by many years. Once the movement disorder is fully established, there is nearly always some degree of cognitive abnormality. Exceptional cases have been reported in which the movement disorder existed for 10 to 30 years without mental changes (Britton et al); this would be most characteristic of patients with fewer CAG repeats. More typically after 10 to 15 years of symptoms, most patients deteriorate to a vegetative state, unable to stand or walk and eating little; in this late stage, a mild amyotrophy may appear. Noteworthy is the high suicide rate, as pointed out by Huntington himself (see also Schoenfeld et al). Because there is a higher-than-normal incidence of head trauma, chronic subdural hematoma is another common finding at autopsy.
The first signs of the disease may appear in childhood, before puberty (even younger than the age of 4 years), and several series of such early-onset cases have been described (Farrer and Conneally; van Dijk et al). Mental deterioration at this early age is more often accompanied by cerebellar ataxia, behavior problems, seizures, bradykinesia, rigidity, and dystonia than by chorea (Byers et al). However, this rigid form of the disease (Westphal variant) also occurs occasionally in adults as mentioned above, in some cases because of HDL2. Functional decline is much faster in children than it is in adults (Young et al).
The dementia is generally more severe in cases of early onset and with correspondingly longer repeat lengths (15 to 40 years of age) than in those of later onset (55 to 60 years of age). In adult patients with early onset, the emotional disturbance tends to be more prominent initially and precedes the chorea and intellectual loss by years; with older age of onset, choreiform features are more often the initial components; in the middle years, dementia and chorea have their onset at nearly the same age. At the other extreme of age, the first features may become evident in the eighties, with orofacial or other dyskinesias that are mistakenly attributed to an exposure to neuroleptic drugs or called “senile chorea” (see Chap. 4).
Gross atrophy bilaterally of the head of the caudate nucleus and putamen is the characteristic abnormality, usually accompanied by a moderate degree of gyral atrophy in the frontal and temporal regions. The caudate atrophy alters the configuration of the frontal horns of the lateral ventricles in that the inferolateral borders do not show the usual bulge formed by the head of the caudate nucleus. In addition, the ventricles are diffusely enlarged (Fig. 39-4); in CT scans, the bicaudate-to-cranial ratio is increased, which corroborates the clinical diagnosis in the moderately advanced case.
Figure 39-4.
Axial CT from a 54-year-old mildly demented woman with a 10-year history of Huntington chorea. The bulge in the inferolateral border of the lateral ventricle, normally created by the head of the caudate nucleus, is obliterated. There is also diffuse enlargement of the lateral ventricles.
The early articles of Alzheimer and Dunlap and the more recent one of Vonsattel and DiFiglia contain the most authoritative descriptions of the microscopic changes. The latter authors have graded the disease into early, moderately advanced, and far advanced stages. In 5 early but genetically verified cases, no striatal lesion was found, which suggests that the first clinical manifestations are based on a biochemical or infrastructural change. This view is supported by the observation that Huntington patients studied with PET show a characteristic decrease in glucose metabolism in the caudate nuclei, which precedes the volumetric loss of tissue (Hayden et al). The striatal degeneration begins in the medial part of the caudate nucleus and spreads, tending to spare the nucleus accumbens. Of the 6 cell types in the striatum (a differentiation based on size, dendritic arborizations, spines, and axon trajectories), the smaller neurons are affected before the larger ones. Loss of dendrites of the small spiny neurons has been an early finding, while the large cells are relatively preserved and exhibit no special alterations.
The anterior parts of the putamen and caudate are more affected than the posterior parts. Some observers have noted changes in the globus pallidus, subthalamic nucleus, red nucleus, cerebellum, and in the pars reticulata of the substantia nigra. In the cerebral cortex, there is slight neuronal loss in layers 3, 5, and 6, with replacement gliosis. Cases are reported with typical striatal lesions but normal cortices in which only chorea had been present during late life. Several neuropathologists have observed marked cell loss and gliosis in the subthalamic nuclei in Huntington-affected children or young adults with chorea and behavior disorders. However, Hadzi and colleagues have determined that the pathologic changes in the striatum and the cortex evolve differently and have separate relationships to the CAG repeat length.
As mentioned above, there is a general relationship between the number of CAG repeats and the age of onset of symptoms. It has been found that it is the longer sequence on either of the 2 alleles that determines the age of onset, the size of the expansion of the normal allele exerting no influence (Lee et al, 2012). Earlier onset in successive generations (anticipation) is well described in the early writings on the subject and is now known to be attributable to increasing lengths of the CAG repeat sequence.
From the molecular perspective, the pathogenesis of this disease is a direct, but still poorly understood, consequence of the aforementioned expansion of the polyglutamine region of huntingtin (the protein product of the Huntington gene). It has been shown that the mutant huntingtin protein aggregates in the nuclei of neurons. Moreover, the protein accumulates preferentially in cells of the striatum and parts of the cortex affected in Huntington disease. Evidence, particularly that given by Wetz (cited in the review by Bates), suggests that these aggregates may be toxic to neurons, either directly or in their protofibrillary (unaggregated) form. The situation is, however, likely to be more complex, as the bulk of huntingtin deposition is found in cortical neurons, whereas the neuronal loss is predominantly striatal. One theory supports the concept that the polyglutamine complex renders certain cell types unduly sensitive to glutamate-mediated excitotoxicity. More recently, 2 mechanisms have been proposed based on an interruption of protein transcription by the binding of mutant huntingtin to transcription proteins or that mitochondrial dysfunction occurs directly or through the same transcriptional mechanism, as summarized by Greenamyre. Because polyglutamine expansions are implicated in several neurodegenerative diseases (reviewed in corresponding sections of this chapter), treatments that block their effects on cellular function may be broadly effective in several degenerative diseases.
Once the disease has been observed in its fully developed form, its recognition requires no great clinical acumen. The main difficulty arises in patients who lack a family history but who display progressive chorea, emotional disturbance, and dementia. This problem has been largely overcome since the mutation was identified. It is now possible to confirm or exclude the diagnosis by analysis of DNA from a blood sample. The presence of more than 39 CAG repeats at the Huntington locus essentially confirms the disease and gives some indication of the expected time of onset; lesser numbers of repeat length leave room for equivocation and strings between 39 and 42 may not be manifest if the patient does not live long enough to express the illness.
Chorea that begins in late life with only mild or questionable intellectual impairment and without a family history of similar disease is a source of diagnostic difficulty. A few cases are the result of the earlier mentioned HDL2 mutation and others derive from alternative degenerative conditions discussed below. Referring to the problem as “senile chorea” does not solve the problem. Indeed, senile chorea has many causes. We have seen it appear with infections, hyperglycemia, drug therapy, strokes, and thyrotoxicosis, only to disappear after a few weeks. A few times we have been confronted with the problem of an older patient who displays orolingual dyskinesias that are most characteristic of exposure to neuroleptic drugs but in whom there was no such history of exposures; testing usually disclosed Huntington disease.
Chorea in early adult life always raises the question of a late form of Sydenham chorea, of lupus erythematosus with antiphospholipid antibodies, or of cocaine use, but neither familial occurrence nor mental deterioration is part of these processes. A “benign inherited chorea,” transmitted as an autosomal dominant trait without prolongation of a triplet sequence, has been traced to chromosome 14q. It is differentiated from Huntington disease by onset before age 5 years, progressing little, and having no associated mental deterioration (Breedveld et al). Other progressive neurologic disorders inherited as autosomal dominant traits and beginning in adolescence or adult life (e.g., polymyoclonus with or without ataxia, acanthocytosis with progressive chorea, and dentatorubropallidoluysian degeneration) can closely mimic Huntington disease, as described further on; sometimes only the genetic and pathologic findings settle the matter. A midlife progressive chorea without dementia (after more than 25 years of followup) that does not display the Huntington genotype has been reported. In at least one family in which this clinical picture is dominantly inherited, the fundamental defect is a mutation in the gene encoding the light chain of ferritin (Curtis). Affected individuals have axonal changes in the pallidum with swollen, ubiquitin- and tau-positive aggregates; serum ferritin levels may be depressed. The implication of this mutation is that perturbations of iron metabolism may be toxic to neurons, a feature that also characterizes Hallervorden-Spatz disease.
Dentatorubropallidoluysian atrophy (DRPLA), sometimes mistaken clinically for Huntington chorea, was described in European families by Warner and associates and is discussed further on. The extrapyramidal manifestations include chorea, myoclonus, and rigidity. Adult-onset chorea and dementia has been described with propionic acidemia; propionic acid is elevated in the plasma, urine, and CSF. This disorder must be added to other metabolic diseases described in Chap. 37 as causes of childhood chorea and dyskinesia—such as glutaric acidemia, keratin sulfaturia, calcification of basal ganglia, phenylketonuria, and Hallervorden-Spatz disease, now called PANK (Hagberg et al).
Other problems in differential diagnosis include prion disease, Wilson disease (see Chap. 37), acquired hepatocerebral degeneration (see Chap. 40), paraneoplastic chorea (see Chap. 31), and most often and especially, tardive dyskinesia (see Chap. 41). Many drugs in addition to the toxic effects of L-dopa and antipsychotic medications occasionally cause chorea (amphetamines, cocaine, tricyclic antidepressants, lithium, isoniazid, linezolid). The hyperglycemic–hyperosmolar state is known for producing a variety of generalized or local movement disorders, prominent among them being chorea.
The dopamine antagonist haloperidol, in daily doses of 2 to 10 mg, is effective partially in suppressing the movement disorder. Because of the danger of superimposing tardive dyskinesia on the chronic disorder, the chorea should be treated only if it is functionally disabling, using the smallest possible dosages. Haloperidol may also help alleviate abnormalities of behavior or emotional lability, but it does not alter the progress of the disease. The authors have not been impressed with the therapeutic effectiveness of other currently available drugs. Levodopa and other dopamine agonists make the chorea worse and, in the rigid form of the disease, evoke chorea. Drugs that deplete dopamine or block dopamine receptors—such as reserpine, clozapine, and particularly tetrabenazine, which has been validated in a controlled study (Huntington Study Group)—suppress the chorea to some degree, but their side effects (drowsiness, akathisia, and tardive dyskinesia) usually outweigh their desired effects. They may be tried in difficult cases. The juvenile (rigid) form of the disease is probably best treated with antiparkinsonian drugs. Preliminary studies of the transplantation of fetal ganglionic tissue into the striatum have achieved mixed results. The psychologic and social consequences of the disease require supportive therapy, and genetic counseling is essential. Antidepression drugs are widely implemented because of the high incidence of depression and suicidality but their efficacy is not clear. Huntington disease pursues a steadily progressive course and death occurs as mentioned, on average 15 to 20 years after onset, sometimes much earlier or later.
There are 2 categories of neurologic disease associated with red blood cell acanthocytosis; one with a defect in the red cell lipid membrane (represented by Bassen-Kornzweig disease and the HARP [hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration] syndrome [see Chap. 37]) and a second group that lacks a lipid abnormality. This latter type of neuroacanthocytosis enters into the differential diagnosis of Huntington chorea or unexplained progressive choreas and has the following characteristics: (1) onset in adolescence or early adult life of generalized involuntary movements (described as chorea but including dystonia and tics), usually beginning as an orofacial dyskinesia and spreading to other parts of the body and to other neural systems; (2) mild to moderate mental deterioration with behavioral disturbance in some but not all cases; (3) decreased or absent tendon reflexes and evidence of chronic axonal neuropathy and denervation atrophy of muscles; and (4) the defining feature of acanthocytosis (thorny or spiky appearance of erythrocytes). The main syndrome, and the one to which the term neuroacanthocytosis had for a long time been applied, is caused by an autosomal recessive mutation. However, there are now 4 additional subtypes, one dominantly transmitted and another X-linked (McLeod type), which is discussed below. These are all in distinction to Bassen-Kornzweig disease that is caused by an inherent defect in the lipid layer of the red cell membrane (see further on).
In the series of 19 cases reported by Hardie and colleagues, the manifestations included dystonia, tics, vocalizations, rigidity, and lip and tongue biting; more than half had cognitive impairment or psychiatric features. The average age of onset was 32 years; 7 of the 19 cases were sporadic. The disease has been linked in almost all families to chromosome 9q, where there is a mutation in the gene encoding a large (3,100-amino-acid) protein designated chorein that is involved in cellular protein sorting and trafficking (Rampoldi). Some of the families with dominantly inherited neuroacanthocytosis have mutations in the chorein gene. There is atrophy and gliosis of the caudate nuclei and putamens but no neuronal loss in the cerebral cortex or other parts of the brain.
According to Sakai and coworkers, the acanthocytosis is the result of an abnormal composition of covalently (tightly) bound fatty acids in erythrocyte membrane proteins (palmitic and docosahexanoic acids increased and stearic acid decreased). The cells should be examined in a fresh preparation of blood and isotonic saline; it is likely to be overlooked in a conventional Wright stain. More than 5 percent of the red cells have the characteristic structural abnormality in affected individuals. The acanthocytosis may also be detected by scanning electron microscopy. The latter may be necessary to undertake in cases of unexplained chorea that have the other features of this disease as genetic testing for the gene (see below) is not widely available.
McLeod disease, another disorder with acanthocytosis and the gradual development of chorea in middle to late life, is characterized by degeneration of the caudate and putamen and a myopathy (elevated serum creatine phosphokinase [CPK]). These individuals have fewer facial tics and orofacial features than those with neuroacanthocytosis. McLeod syndrome arises from mutations in a gene on the X-chromosome that encodes the KX protein, which binds to surface Kell antigens on red cells. In addition to the primary KX gene mutations, these individuals show diminished Kell antigen expression on the red-cell surface.
Included in this category are a heterogeneous group of degenerative diseases in which the symptoms of parkinsonism and corticospinal degeneration are present in various combinations. Some of the diseases that make up this group have not been sharply delineated and are difficult to separate from one another.
Variants of this category of disease continue to appear, all rare. The authors have observed several patients in whom extreme rigidity, corticospinal signs but no dementia, have developed over a period of several years. In the later stages of the disease, the patient, while alert, is totally helpless and unable to speak, swallow, or move the limbs. Only eye movements are retained, and even these are hampered by supranuclear gaze palsies in advanced cases. Intellectual functioning appears to be better preserved than movement but is difficult to assess. Other bodily functions are intact. The course is slowly progressive and ends fatally in 5 to 10 years. There is no family history of similar disease, and there are no clues as to causation. Gilbert and colleagues have described similar cases with signs of Parkinson disease, motor neuron disease, and dementia; in their cases, there were no senile plaques or Lewy bodies. The concurrence of typical motor neuron disease and Parkinson disease may be coincidental, but Qureshi and colleagues described 13 patients in whom both clinical phenomena began within a short time and they considered them to be related. In the variant described by Tandan and colleagues, an autosomal dominant syndrome of Charcot-Marie-Tooth polyneuropathy was combined with ptosis, parkinsonism, and dementia, again without Lewy bodies or amyloid plaques. Other variants have been described by Schmitt and coworkers and by Mata and colleagues. Hudson reviewed 42 sporadic cases in which ALS-parkinsonism-dementia were combined.
Under the title “Spastic Pseudosclerosis,” Jakob, in 1921, described a chronic disease of middle to late adult life, characterized by abnormalities of behavior and intellect; weakness, ataxia, and spasticity of the limbs (chiefly the legs); extrapyramidal symptoms such as rigidity, slowness of movement, tremors, athetotic postures, and hesitant, dysarthric speech; and normal spinal fluid. The pathologic changes were diffuse and consisted mainly of an outfall of neurons in the frontal, temporal, and central motor gyri, striatum, ventromedial thalamus, and bulbar motor nuclei. In one of Jakob’s cases, there were also prominent changes in the anterior horn cells and corticospinal tracts in the spinal cord like those of ALS. The latter finding gave rise to Wilson’s concept of the disease as a corticostriatospinal degeneration. Some restricted cases bear a resemblance to the type of frontotemporal dementia that occurs with motor neuron disease.
A degenerative and probably familial disorder that had been described earlier by Creutzfeldt was considered by Spielmeyer to be sufficiently similar to the one of Jakob to warrant the designation Creutzfeldt-Jakob disease. As discussed in Chap. 33, the disorder originally described by Creutzfeldt and Jakob has been a source of endless controversy because of its indeterminate character. It has been confused with the subacutely evolving myoclonic dementia, or subacute spongiform encephalopathy, which is now known to be an infection caused by a prion agent. The latter disease bears at best only a superficial resemblance to the one described by Creutzfeldt and Jakob, and the 2 disorders should be separated. Unfortunately, the use of the eponym for the prion-related disease is so entrenched that attempts to delete it are futile and probably unnecessary. However, the term Jakob disease has been used for the degenerative type of corticostriatospinal degeneration.
The Guamanian Parkinson-dementia-ALS complex deserves separate comment because there have been many carefully studied cases with almost uniform clinical and pathologic features. The disease occurs in the indigenous Chamorro peoples of Guam and the Mariana islands, predominantly in men between the ages of 50 and 60 years. Progressive parkinsonism and dementia are combined with upper or lower motor neuron disease (ALS is also common among the Chamorro) leading to death in 5 years. The pathologic changes, described by Hirano and associates, consist of severe cortical atrophy with neurofibrillary tangles and a depopulation of the substantia nigra, but notably no Lewy bodies or amyloid plaques, even with sensitive neurochemical staining. Cases with amyotrophy show a loss of anterior horn cells. The cause of the Guamanian multisystem degeneration is not known, although several studies have incriminated one or more putative neurotoxins in the food supply (see Chap. 43). There are some clinical and pathologic similarities to the form of frontotemporal dementia with motor neuron disease.
Occasionally, the authors have encountered families in which several members developed a spastic paraparesis and a gradual failure of intellectual function during the middle adult years. The patient’s mental horizon narrowed gradually, and the capacity for high-level thinking diminished; in addition, the examination showed exaggerated tendon reflexes, clonus, and Babinski signs. In one such family, the illness had occurred in 2 generations; in another, 3 brothers in a single generation were afflicted. Skre described 2 recessive types of hereditary spastic paraplegia in Norway, 1 with onset in childhood, the other with onset in adult life. In contrast to the dominant form (see further on), the recessive types displayed evidence of more widespread involvement of the nervous system, including dementia, cerebellar ataxia, and epilepsy. Also, Cross and McKusick have observed a recessive type of paraplegia accompanied by dementia beginning in adolescence. They named it the Mast syndrome, after the afflicted family.
Worster-Drought and others reported the pathologic findings in 2 cases of this type. In addition to plaques and neurofibrillary changes, there was demyelination of the subcortical white matter and corpus callosum and a “patchy but gross swelling of the arterioles,” which gave the staining reactions for amyloid (“Scholz’s perivascular plaques”). van Bogaert and associates published an account of similar cases that showed the characteristic pathologic features of Alzheimer disease.
Another interesting association of familial spastic paraplegia is with progressive cerebellar ataxia. Fully one-third of the cases that we have seen with such a spastic weakness were also ataxic and would fall into the category of spinocerebellar degenerations. Yet another variant of this group of diseases has been described by Farmer and colleagues; the inheritance in their cases was autosomal dominant, and the main clinical features were deafness and dizziness, ataxia, chorea, seizures, and dementia, evolving in that order. Postmortem examinations of 2 patients disclosed calcification in the globus pallidus, neuronal loss in the dentate nuclei, and destruction of myelinated fibers in the centrum semiovale.
Under this title, Robitaille and colleagues have described a progressive neurologic disease in adults characterized clinically by spasticity, chorea, dementia, and a predominantly sensory polyneuropathy that is reviewed in more detail in Chap. 39. Structures that closely resembled Lafora bodies and corpora amylacea were found in large numbers in both central and peripheral neural processes (mainly in axons) and also in astrocytes. These basophilic PAS-positive structures were composed of glucose polymers (polyglucosans) and were readily demonstrated in sural nerve biopsies and therefore probably best termed polyglucosan bodies. Some of these structures were also found in the heart and liver.
More recently, Rifal and associates reviewed the findings in 25 cases of this disease—one observed by them and 24 reported previously. The dementia was relatively mild, consisting of impairment of retentive memory, dysnomia, dyscalculia, and sometimes nonfluent aphasia and deficits of “visual integration”; this was overshadowed by rigidity and spasticity of the limbs and the peripheral nerve disorder. Bladder dysfunction has been an early sign in many patients including a middle-aged woman under our care who had only diffuse white matter changes in the cerebral MRI and a moderate sensory neuropathy. Nerve conduction velocities were diminished and the leg muscles were denervated. Moderate degrees of generalized cerebral atrophy, multifocal areas of white matter rarefaction, and degeneration of the corticospinal system, disclosed by MRI. Some cases simulate motor neuron disease. The finding of polyglucosan axonal inclusion in biopsied nerves confirms the diagnosis. The disease has sometimes been misinterpreted as adrenoleukodystrophy. The disorder appears to be a glycogenosis that is allied with Anderson disease, as discussed in Chaps. 38 and 48.
Adult forms of metachromatic leukodystrophy, adrenoleukodystrophy, Krabbe disease, and neuronal ceroid lipofuscinosis (Kufs disease) may be present with a similar clinical picture of progressive dementia (see Chap. 37) as may Whipple disease or the Wernicke-Korsakoff disease. Quite rare instances of the same syndrome with adult onset have proved to be caused by phenylketonuria or other aminoacidopathies (see Chap. 37).
Diseases Characterized by Abnormalities of Posture and Movement
This common disease, known since ancient times, was first cogently described by James Parkinson in 1817. In his words, it was characterized by “involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forward, and to pass from a walking to a running pace, the senses and intellect being uninjured.” Strangely, his essay contained no reference to rigidity or to slowness of movement and it stressed unduly the reduction in muscular power. The same criticism can be leveled against the term paralysis agitans, which appeared for the first time in 1841 in Marshall Hall’s textbook Diseases and Derangements of the Nervous System and has fallen out of use, but was such a common term in the literature that it is included here.
The natural history of the disease is of interest. As a rule, it begins between 45 and 70 years of age, with the peak age of onset in the sixth decade. It is infrequent before 30 years of age, and most series contain a somewhat larger proportion of men. Trauma, emotional upset, overwork, exposure to cold, “rigid personality,” and so on, were among many factors that had been suggested over the years as predisposing to the disease, but there is no evidence to support any such claims. Idiopathic Parkinson disease is observed in all countries, all ethnic groups, and all socioeconomic classes, although the incidence in African Americans is only one-quarter that in whites. There may be an increased incidence in rural compared to urban areas. In Asians, the incidence is one-third to one-half that in whites. The disease is frequent in North America, where there are approximately 1 million affected patients, constituting about 1 percent of the population over the age of 65 years. The incidence in European countries where vital statistics are kept is similar. A possible relationship to repeated cerebral trauma and to the “punch-drunk” syndrome (dementia pugilistica; chronic traumatic encephalopathy) has been particularly problematic and is unresolved despite several celebrated cases (Lees). A protective effect of smoking and coffee drinking has emerged in some epidemiologic studies but is marginal.
A tetrad of hypo- and bradykinesia, resting tremor, postural instability, and rigidity are the core features of Parkinson disease. These are evident as an expressionless face, poverty and slowness of voluntary movement, “resting” tremor, stooped posture, axial instability, rigidity, and festinating gait. Much can still be gained from perusal of the often-cited study by Hoehn and Yahr, published in 1967 before the widespread use of L-dopa. Table 39-2 is reproduced from that paper. The manifestations of basal ganglionic disease are fully described in Chap. 4, and only certain diagnostic problems and variations of the clinical picture need be considered here.
Tremor | 70% |
Gait disturbance | 11% |
Stiffness | 10% |
Slowness | 10% |
Muscle aches | 8% |
Loss of dexterity | 7% |
Handwriting disturbance | 5% |
Depression, nervousness, other psychiatric disturbance | 4% |
Speech disturbance | 3% |
The early symptoms may be difficult to appreciate and are often overlooked by family members because they evolve slowly and tend to be attributed to the natural changes of aging. Speech becomes soft, monotonous, and cluttered. For a long time the patient may not be conscious of the inroads of the disease. At first the only complaints may be of aching of the back, neck, shoulders, or hips and of vague weakness. Slight stiffness and slowness of movement or a reduction in the natural swing of one arm during walking are ignored until one day it occurs to the physician or to a member of the family that the patient has the overall cast of Parkinson disease. Infrequency of blinking, as originally pointed out by Pierre Marie, is an early sign. The usual blink rate (12 to 20/min) is reduced in the parkinsonian patient to 5 to 10/min, and with it there is a slight widening of the palpebral fissures, creating a stare. A reduction in movements of the small facial muscles imparts the characteristic expressionless “masked” appearance (hypomimia). When seated, the patient makes fewer small shifts and adjustments of position than the normal person (hypokinesia), and the fingers straighten and assume a flexed and adducted posture at the metacarpophalangeal joints.
The characteristic tremor, which usually involves a hand, is often listed as the initial sign; but in at least half the cases observant family members will already have remarked on the patient’s relative slowness of movement. In about one-quarter of cases the tremor is mild and intermittent, or evident in only one finger or one hand. The tremor of the fully developed case takes several forms, as was remarked in Chap. 6. The 4-per-second “pill-rolling” tremor of the thumb and fingers, although most characteristic, is seen in only about half the patients. It is typically present when the hand is motionless, that is not used in voluntary movement (hence the commonly used term resting tremor). Complete relaxation, however, reduces or abolishes the tremor, so that the term tremor in the position of repose is actually a more accurate description. Volitional movement dampens it momentarily. The rhythmic beat coincides with an alternating burst of activity in agonist and antagonist muscles in the electromyogram (EMG); hence the description alternating tremor is applied. The arm, jaw, tongue, eyelids, and foot are less often involved. Even the least degree of tremor is felt during passive movement of a rigid part (cogwheel phenomenon, or Negro sign, or at least this is the ostensible explanation for cogwheeling). The tremor shows surprising fluctuations in severity and is aggravated by walking and excitement, but its frequency remains constant (Hunker and Abbs). It bears repetition that one side of the body is typically involved before the other with tremor and rigidity, and the tremor in particular remains asymmetrical as the illness advances.
Lance and associates have called attention to the high incidence of a second essential type of tremor in Parkinson disease—a fine, 7- to 8-per-second, slightly irregular, action tremor of the outstretched fingers and hands. This tremor, unlike the slower one, persists throughout voluntary movement, is not evident with the limb in a resting position, and is more easily suppressed by relaxation. Electromyographically, it lacks the alternating bursts of action potentials seen in the typical tremor and resembles, if not equates with, essential tremor (see Table 6-1). It is subject to modulation by different medications than those used for the alternating Parkinson tremor. The patient may have either type of tremor or both.
Rigidity is less often an early finding. Once rigidity develops, it is constantly present and can be felt by the palpating fingers and as a salience of muscle groups even when the patient relaxes. When the examiner passively moves the limb, a mild resistance appears from the start (without the short free interval that characterizes spasticity) and it continues evenly throughout movement in both flexor and extensor groups, being interrupted to a variable degree only by the cogwheel phenomenon. Rigidity and its cogwheel component are elicited or enhanced by having the patient engage the opposite limb in a motor task requiring some degree of concentration, such as tracing circles in the air (termed Froment sign, or Noïka-Froment sign when the patient is asked to raise the other arm as high as possible, but this maneuver was actually utilized first to bring out cogwheeling in essential tremor) or touching each finger to the thumb. In the muscles of the trunk, postural hypertonus predominates in the flexor groups and confers on the patient the characteristic flexed posture. Other particulars of the parkinsonian appearance of muscle tone, stance, and gait are discussed in detail in Chaps. 4 and 7. There should be no pyramidal signs in Parkinson disease.
Here, a few additional points should be made regarding the quality of volitional and postural movements. The patient is slow and ineffective in attempts to deliver a quick hard blow; he cannot complete a rapid (ballistic) movement. On the EMG, the normal single burst of agonist–antagonist–agonist sequence of energizing activity is replaced by several sequential brief bursts, according to Hallett and Khoshbin. Alternating movements, at first successful, become progressively impeded if performed repetitively and, finally, they are blocked completely or adopt the rhythm of the patient’s alternating tremor. The patient has great difficulty in executing 2 motor acts simultaneously. In the past the impaired facility of movement had been attributed to rigidity, but the observation that certain surgical lesions in the brain abolished rigidity without affecting movement refuted this interpretation. Thus slowness and lack of natural movements (bradykinesia and hypokinesia, respectively) are not derived from rigidity but are independent manifestations of the disease. The bradykinetic deficits underlie the characteristic poverty of movement, reflected also by infrequency of swallowing, slowness of chewing, a limited capacity to make postural adjustments of the body and limbs in response to displacement of these parts, a lack of small “movements of cooperation” (as in arising from a chair without first adjusting the feet), absence of arm swing in walking, and most of the other aspects of the parkinsonian countenance. Despite a perception of muscle weakness, the patient is able to generate normal or near-normal power, especially in the large muscles; however, in the small ones, strength is slightly diminished.
As the disorder of movement worsens, all customary activities show the effects. Handwriting becomes small (micrographia), tremulous, and cramped, as first noted by Charcot. Speech softens and seems hurried, monotonous, and mumbling (cluttered): The voice becomes less audible and, finally, the patient only whispers. Caekebeke and coworkers refer to the speech disorder as a hypokinetic dysarthria and attribute it to combined respiratory, phonatory, and articulatory dysfunctions. There is a failure to fully close the mouth. The consumption of a meal takes an inordinately long time. Each morsel of food must be swallowed before the next bite is taken.
Walking becomes reduced to a shuffle; the patient frequently loses balance, and in walking forward or backward seems to be “chasing” the body’s center of gravity with a series of increasingly rapid short steps in order to avoid falling (festination). Defense and righting reactions are faulty. Falls do occur, but surprisingly infrequently given the degree of postural instability. Gait is improved by sensory guidance, as by holding the patient at the elbow. Obstacles such as door thresholds have the opposite effect, at times causing the patient to “freeze” in place. Getting in and out of a car or elevator or walking into a room or in a hall becomes particularly difficult. Difficulty in turning over in bed is a similarly characteristic feature as the illness advances, but the patient rarely volunteers this information. Several of our patients have fallen out of bed at a frequency that suggests a connection to their reduced mobility combined with slowed corrective or defensive postural movements. Shaving or applying lipstick becomes difficult, as the facial muscles become more immobile and rigid.
Persistent extension or clawing of the toes, jaw clenching, and other fragments of dystonia, often quite painful, may enter the picture and are sometimes early findings. (These are particularly resistant to treatment.) A special problem of camptocormia occurs in some Parkinson patients wherein an extreme forward flexion of the spine and correspondingly severe stooping occur. It appears to be a type of axial dystonia when it occurs with Parkinson disease. The deformity resolves when the patient is supine or pushes upward on the handles of a walker. This symptom is associated with a variety of other diseases, some of them muscular. We have not been impressed that it is ameliorated by L-dopa. Why some patients with Parkinson disease are extremely bent over and others are not at all affected is unknown.
As noted above, these various motor impediments and tremors characteristically begin in one limb (more often the left) and spread to one side and later to both sides until the patient is quite helpless. Yet in the excitement of some unusual circumstance (as escaping from a fire, for example), the patient with all but the most advanced disease is capable of brief but remarkably effective movement (kinesis paradoxica).
Regarding elicitable neurologic signs, there is an inability to inhibit blinking in response to a tap over the bridge of the nose or glabella (Myerson sign) but grasp and suck reflexes are not present unless dementia supervenes and buccal and jaw jerks are rarely enhanced. Commonly there is an impairment of upward gaze and convergence; if prominent or noted early in the disease, this sign suggests more the possibility of progressive supranuclear palsy. Bradykinesia may extend to eye movements, in that there is a delay in the initiation of gaze to one side, slowing of conjugate movements (decreased maximal saccadic velocity), hypometric saccades, and breakdown of pursuit movements into small saccades.
There are no sensory findings, but a wide variety of paresthetic and other sensory complaints and discomforts are common. These affect mainly the calves and abdomen and are among the most distressing of the nonmotor parkinsonian symptoms. Drooling is troublesome; an excess flow of saliva has been assumed, but actually the problem is probably one of failure to swallow with normal frequency. Seborrhea and excessive sweating are claimed to be secondary as well, the former due to failure to cleanse the face sufficiently, the latter to the effects of the constant motor activity but this explanation seems lacking to us; an autonomic disturbance is more plausible. Other nonmotor features are mostly in the category of autonomic disturbances and include most prominently constipation, abdominal pains and cramps, erectile dysfunction, joint aches, and various other sensory experiences that may be difficult for the patient to describe. There is a tendency in some patients to have orthostatic hypotension and sometimes syncope; this has been attributed by Rajput and Rozdilsky to cell loss in the sympathetic ganglia. However, these features are not as prominent as in multiple system atrophy (Shy-Drager syndrome). It is worth mentioning that several of our younger Parkinson patients with recurrent syncope proved to have cardiac arrhythmias; hence other causes of fainting must be considered.
Postural instability is a core feature of the illness; it can be elicited by tugging at the patient’s shoulders from behind and noting the lack of a small step backward to maintain balance often with a fall or the initiation of backward festination. The tendon reflexes vary, as they do in normal individuals from being barely elicitable to brisk. Even when parkinsonian symptoms are confined to one side of the body, the reflexes are usually equal on the two sides, and the plantar responses are flexor. Exceptionally, the reflexes on the affected side are slightly brisker, which raises the question of corticospinal involvement, but the plantar reflex remains flexor. In these respects, the clinical picture differs from that of corticobasal ganglionic degeneration, in which rigidity, hyperactive tendon reflexes, and Babinski signs are combined with apraxia (see further on).
As mentioned earlier, Parkinson disease may be complicated by dementia, a feature described by Charcot. The reported frequency of this combination varies considerably based on the selection of patients and type of testing. An estimate of 10 to 15 percent (Mayeux et al) is the generally accepted figure and matches our experience. The incidence increases with advancing age and duration of disease, approaching 65 percent in Parkinson patients older than 80 years of age, but mental decline may become apparent in patients in their late fifties. The pathologic basis of the dementia is discussed below.
The overall course of the disease is quite variable. In the majority of patients, the mean period of time from inception of the disease to a chairbound state is 7.5 years, but with a wide range (Hoehn and Yahr; Martilla and Rinne). As much as 10 percent of cases remain relatively mild and only very gradually progressive, and such patients may remain almost stable for 10 years or more. These trajectories have been altered somewhat by modern therapies.
Mentioned here is a rare syndrome described by Klawans and elaborated in a series of 30 patients by Wijemanne and Jankovic. The typical case shows atrophy in one or more body parts, including at times the face, often since childhood, and usually quite subtle. Signs of progressive parkinsonism or dystonia begin in midlife on the atrophic side and, for the most part, are responsive to L-dopa, but some, such as Klawans’ original patients, are resistant. Several types of early life cerebral injury underlie the syndrome, but half of patients have no such lesion evident. Understanding of the idiopathic cases is limited. Those with deep brain lesions may be experiencing a slow degeneration of basal ganglia pathways.
The 2 main difficulties are to distinguish typical Parkinson disease from the many parkinsonian syndromes caused by other degenerative conditions and by medications or toxins, and to distinguish the Parkinson tremor from other types, especially essential tremor. It is worth noting that Parkinson disease is far more common than any of the degenerative syndromes that resemble it. Bradykinesia and rigidity of the limbs and axial musculature are symptoms shared with other forms of parkinsonism, but it is mainly in Parkinson disease that one observes an early sign of “resting” alternating tremor that is more prominent in one arm.
When not all the typical signs are evident, there is no alternative but to reexamine the patient at several-month intervals until it is clear that Parkinson disease is present or until the characteristic features of another degenerative process become evident; these include early falls and vertical gaze impairment in progressive supranuclear palsy; dysautonomia with fainting, bladder, or vocal cord dysfunction in multiple system atrophy; early and rapidly evolving dementia or intermittent psychosis in Lewy-body disease; or apraxia in corticobasal ganglionic degeneration. Very symmetrical findings, particularly tremor, suggest an alternative to idiopathic Parkinson disease. Also, the constellation of features termed “lower half parkinsonism” consisting of difficulty purely with gait and stability, as discussed below and in Chap. 7, suggest a process other than Parkinson disease.
If the symptoms warrant, a beneficial and sustained response to levodopa or a dopamine agonist also gives a reasonably secure, although not entirely conclusive, indication of the presence of Parkinson disease (see further on). The other parkinsonian syndromes are for the most part changed only slightly or only for a few weeks or months by the drug. Conversely, although some experts disagree, we have adhered to the notion that complete resistance of the symptoms to L-dopa early in the illness makes the diagnosis unlikely. Furthermore, almost all patients with idiopathic Parkinson disease eventually acquire dyskinesias in response to L-dopa and the absence of this sign after approximately 3 to 5 years of use of the drug brings the diagnosis into question.
The epidemic of encephalitis lethargica (von Economo encephalitis) that spread over Western Europe and the United States after the First World War left great numbers of parkinsonian cases in its wake. No definite instance of this form of encephalitis had been recorded before the period 1914 to 1918, and very few have been seen since 1930; hence, this type of postencephalitic parkinsonism is no longer a diagnostic consideration. However, a Parkinson-like syndrome has been described following other forms of encephalitis, particularly with Japanese B virus, West Nile virus, and eastern equine encephalitis. In the few cases caused by these viruses that we have observed, there has been fairly symmetrical rigidity, hypokinesia, and little or no tremor.
An “arteriopathic” or “arteriosclerotic” form of Parkinson disease was at one time much diagnosed but we have never been entirely convinced of its reality, referring to damage to the substantia nigra as a result of vascular disease or to a syndrome that closely resembles Parkinson disease as a result of atherosclerotic white matter damage. Nonetheless, a number of authoritative clinicians are of the opinion that patients with a vascular cause have a predominantly “lower half” parkinsonism in which shuffling gait, stickiness on turning, and falling are disproportionate to other features. There is no tremor, and little or no response to L-dopa (see Winikates and Jankovic). MRI in such cases has shown substantial white matter changes in both cerebral hemispheres. In the few cases attributable to vascular parkinsonism that have come to our attention with autopsy material, there have been Lewy bodies in the appropriate locations. Pseudobulbar palsy from a series of lacunar infarcts or from Binswanger disease can cause a clinical picture that simulates certain aspects of Parkinson disease, but unilateral and bilateral corticospinal tract signs, hyperactive facial reflexes, spasmodic crying and laughing, and other characteristic features distinguish spastic bulbar palsy from Parkinson disease. Of course, the elderly parkinsonian patient is not impervious to cerebrovascular disease, and the 2 conditions overlap, but differentiating the predominantly gait or dementing disorders of widespread vascular brain damage from idiopathic Parkinson disease is not difficult.
Normal-pressure hydrocephalus can undoubtedly produce a syndrome resembling Parkinson disease, particularly in regard to gait and postural instability, and at times extending to bradykinesia; but rigid postures, slowness of alternating movements, hypokinetic ballistic movements, and resting tremor are not part of the clinical picture. The gait tends to be short-stepped but not shuffling and there is more of a tendency to retropulsion than there is in Parkinson disease. Sometimes a lumbar puncture gives surprising benefit, indicating hydrocephalus as the cause of the motor slowing and gait disorder.
Essential tremor is distinguished by its fine, quick quality, its tendency to become manifest during volitional movement and to disappear when the limb is in a position of repose, and the lack of associated slowness of movement or of flexed postures. Cogwheeling of minor degree may be associated. The head and voice are more often truly tremulous in essential tremor than in Parkinson disease. Some of the slower, alternating forms of essential tremor are difficult to distinguish from parkinsonian tremor; one can only wait to see whether it is the first manifestation of Parkinson disease. A markedly asymmetrical or unilateral tremor favors Parkinson disease. Also as noted, a faster oscillation is often mixed with the slow alternating Parkinson tremor, but the fast-frequency tremor is only occasionally an opening feature of the disease as discussed in Chap. 6.
Progressive supranuclear palsy (discussed in a section further on) is characterized by rigidity and dystonic postures of the neck and shoulders, a staring and immobile countenance, and a tendency to topple when walking—all of which are vaguely suggestive of Parkinson disease. Early and frequent falls are particularly suggestive of this disease, not being atypical of Parkinson disease until its late stages. Inability to produce vertical saccades and, later, paralysis of upward and downward gaze and eventual loss of lateral gaze with retention of reflex eye movements establish the diagnosis of PSP in most cases.
Paucity of movement, unchanging attitudes and postural sets, and a slightly stiff and unbalanced gait may be observed in patients with an anergic or hypokinetic type of depression. Because a fair proportion of parkinsonian patients are depressed, the separation of these 2 conditions is at times difficult. The authors have seen patients who were called parkinsonian by competent neurologists but whose movements became normal when antidepressant medication or electroconvulsive therapy was given. Several such patients have nonetheless insisted that levodopa helps them in some nondescript way.
The rapid onset of parkinsonism should suggest exposure to neuroleptic medications (used at times as antiemetics and gastric motility agents [metoclopramide]), a variant of Creutzfeldt-Jakob disease, an unusual postinfectious or paraneoplastic illness, or viral encephalitis. The implicated drugs may also evoke an inner restlessness, a “muscular impatience,” an inability to sit still, and a compulsion to move about much like that which occurs at times in the parkinsonian patient (akathisia). Even the newer antipsychosis medications, favored specifically because of a putative lack of extrapyramidal effects, may be at fault.
Strict adherence to the diagnostic criteria for Parkinson disease also permits its differentiation from corticostriatospinal, striatonigral, and corticobasal ganglionic degeneration, calcification of the basal ganglia, Wilson disease, the acquired hepatolenticular degeneration of repeated hepatic coma, manganese poisoning, as well as Machado-Joseph disease, all of which are discussed in other parts of this chapter.
All in all, if one adheres to the standard definition of Parkinson disease—bradykinesia, hypokinesia “resting” tremor, postural changes and instability, cogwheel rigidity, and response to L-dopa—errors in diagnosis are few. Yet in a series of 100 cases, studied clinically and pathologically by Hughes and associates, the diagnosis was inaccurate in 25 percent. The ostensible explanation for this difficulty is that approximately one-quarter of Parkinson patients fail to display the characteristic tremor and approximately 10 percent are said to not respond to L-dopa. These authors noted that early dementia and autonomic disorder and the presence of ataxia or corticospinal signs were reliable guides to an alternate diagnosis.
The most constant and pertinent finding in both idiopathic and postencephalitic Parkinson disease is a loss of pigmented cells in the substantia nigra and other pigmented nuclei (locus ceruleus, dorsal motor nucleus of the vagus). The substantia nigra is visibly pale to the naked eye; microscopically, the pigmented nuclei show a marked depletion of cells and replacement gliosis, and some of the remaining cells have reduced quantities of melanin, findings that enable one to state with confidence that the patient must have suffered from Parkinson disease. Also, many of the remaining cells of the pigmented nuclei contain eosinophilic cytoplasmic inclusions, surrounded by a faint halo, called Lewy bodies (Fig. 39-5). These are seen in practically all cases of idiopathic Parkinson disease. They were generally absent in postencephalitic cases, but there were neurofibrillary tangles within nigral cell in that disorder. Both these cellular abnormalities appear occasionally in the substantia nigra of aged, nonparkinsonian individuals. Possibly the individuals with Lewy bodies would have developed Parkinson disease had they lived a few more years. Many of the inherited forms of Parkinson disease also lack Lewy bodies.
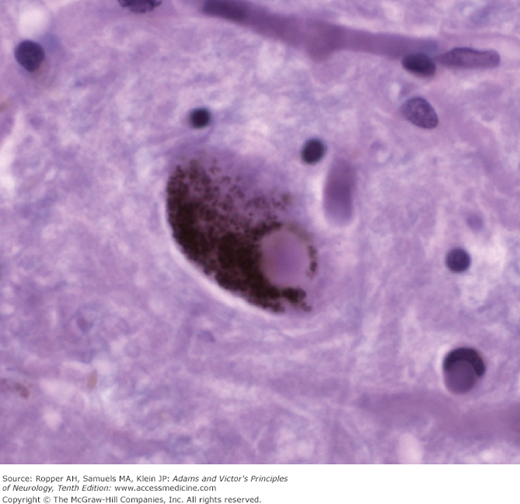
Noteworthy is the finding by McGeer and colleagues that nigral cells normally diminish with age, from a maximal complement of about 425,000 to 200,000 at age 80 years. Tyrosine-hydroxylase, the rate-limiting enzyme for the synthesis of dopamine, diminishes correspondingly. However, these authors and others have found that in patients with Parkinson disease the number of pigmented neurons is reduced to 30 percent or less of that in age-matched controls. Using more refined counting techniques, Pakkenberg and coworkers estimated the average total number of pigmented neurons to be 550,000 and to be reduced in absolute numbers by 66 percent in Parkinson patients. (The number of nonpigmented neurons was reduced in Parkinson cases by only 24 percent.) Thus aging contributes importantly to nigral cell loss, but the cell depletion is so much more marked in Parkinson disease that some factor other than aging must also be operative.
Other regions of neuronal loss are widespread as mentioned, but their significance is less clear. There is neuronal loss in the mesencephalic reticular formation, near the substantia nigra. These cells project to the thalamus and limbic lobes. In the sympathetic ganglia, there is slight neuronal loss and Lewy bodies are seen. This is also true of the pigmented nuclei of the lower brainstem as well as of neuronal populations in the putamen, caudatum, pallidum, and substantia innominata. On the other hand, dopaminergic neurons that project to cortical and limbic structures, to caudate nucleus and nucleus accumbens, and to periaqueductal gray matter and spinal cord are affected little or not at all. The lack of a consistent lesion in either the striatum or the pallidum is noteworthy. An alternative hypothesis offered by Braak and Tredici, mentioned in an earlier section of this chapter and attributed to Braak and Braak, is that the substantia nigra compacta is affected only late in the pathobiology of Parkinson disease. Their study found that the earliest changes in the brain occur in the dorsal glossopharyngeal-vagal and anterior olfactory nuclei, and only later did they appear in the midbrain nuclei. This theory accommodates a variety of clinical features and potential environmental triggers to the disease. Lang has suggested that this distribution of cell loss explains some of the nondopaminergic features of the disease and offers other avenues for therapy.
Related posts:
 Chapter 6. Tremor, Myoclonus, Focal Dystonias, and Tics
Chapter 18. Faintness and Syncope
Chapter 15. Deafness, Dizziness, and Disorders of Equilibrium
Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 35. Craniocerebral Trauma
Chapter 52. Depression and Bipolar Disease
Chapter 6. Tremor, Myoclonus, Focal Dystonias, and Tics
Chapter 18. Faintness and Syncope
Chapter 15. Deafness, Dizziness, and Disorders of Equilibrium
Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 35. Craniocerebral Trauma
Chapter 52. Depression and Bipolar Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


