Electrophysiologic and Laboratory Aids in the Diagnosis of Neuromuscular Disease: Introduction
The clinical suspicion of neuromuscular disease, disclosed by any of the symptoms or syndromes in the succeeding chapters, finds ready confirmation in the laboratory. The intelligent selection of ancillary examinations requires knowledge of the biochemistry and physiology of nerve action potentials, neuromuscular transmission, and muscle fiber contraction. These basic subjects, with the relevant anatomy, serve as an introduction to the descriptions of the laboratory methods and the subject matter of the chapters on the diseases of muscle and nerve that follow.
Electrolytes and Neuromuscular Activity
Since the early studies of Hodgkin (1951) and of Hodgkin and Huxley (1952), tomes have been written on the subject of the conduction of electrical impulses in neural tissues. Such conduction in nerve and muscle depends first on the maintenance of a fluid internal environment that is distinctly different from the external or interstitial medium. The main intracellular constituents are potassium (K), magnesium (Mg), and phosphorus (P), whereas those outside the cell are sodium (Na), calcium (Ca), and chloride (Cl). In both nerve and muscle the intracellular concentrations of these ions are held within a narrow range by both passive electrical and active chemical forces, which maintain the membranes in an electrochemical equilibrium termed the resting membrane potential. These forces are the result of selective permeability of the membranes to various ions and to the continuous expulsion of intracellular Na through specific channels by a pump mechanism (the sodium pump). The function of the sodium pump is dependent on the enzyme Na-K-ATPase (adenosine triphosphatase), which is localized in the membranes.
This resting membrane potential is the result of the differential concentrations of K and Na. The interior of the cell is some 30 times richer in K than the extracellular fluid, and the concentration of Na is 10 to 12 times greater in the extracellular fluid. In the resting state, the chemical forces that promote diffusion of K ions out of the cell (down their concentration gradient) are counterbalanced by electrical forces, the internal negativity, that opposes further diffusion of K to the exterior of the cell. The situation of Na ions in the equilibrium state is the opposite; they tend to diffuse into the cell, both because of their concentration gradient and because of the relative negativity inside the cell. Because the membrane at rest is less permeable to Na than to K, the amount of K leaving the cell exceeds the amount of Na entering the cell, thus creating the difference in electrical charge across the membrane. This discrepancy creates an electrical potential across the membrane such that, in the resting state, the intracellular compartment is 70 to 90 mV negative relative to the extracellular space (the resting membrane potential). The actions of the Na:K pump and the presence of impermeant, negatively charged intracellular proteins, contribute to maintaining this negative potential.
From this resting potential, any electrical discharge of neural and muscular tissue is predicated on the special property of excitable membranes; namely, the permeability of the cell to Na is linked to the electrical potential across the membrane by voltage-gated (i.e., voltage sensitive or voltage triggered) ion channels. Any depolarization of the membrane by a slight electrical or chemical change, creates an increased permeability to Na through the sodium channels and a consequent inward movement of sodium. Subsequently, K moves outward and repolarizes the membrane and thereby reduces its permeability to Na. If a greater degree of depolarization occurs, a situation arises in which the outward movement of K is unable to stabilize the membrane potential. The membrane then becomes even more depolarized and progressively more permeable to Na, and an “explosive,” or regenerative Na current develops in which Na rushes down its chemical and electrical gradients into the cell. With the elevation in Na permeability, the Na ions continue to depolarize the intracellular compartment to about +40 mV (the Nernst equilibrium potential for Na). It is this depolarization of the intracellular compartment, lasting a few milliseconds, that constitutes the action potential.
However, an equilibrium at this level is not established because there is a rapid (within 5 ms) fall in membrane Na permeability back to baseline levels. This fall in Na permeability occurs because the depolarization inactivates Na conductance. The transient depolarization at the same time activates an increase in membrane permeability to K. The combined effects of the resulting efflux of K and a diminishing influx of Na repolarizes the membrane back to its resting level. During a brief period immediately after repolarization, the nerve and muscle fibers are refractory, at first absolutely then relatively, to another depolarizing stimulus. The duration of the refractory period is determined largely by the duration of the inactivation of the Na channel.
Conduction, or propagation, of the action potential along nerve or muscle, occurs as the current created by depolarization of a small segment of membrane flows into the contiguous membrane, which, in turn, becomes depolarized. When the depolarization reaches the threshold for development of an action potential, a new zone of increased Na permeability is created. In this manner, the action potential spreads in an “all-or-none” fashion, down the length of the nerve or muscle membrane. The action potentials in individual nerve fibers are too small and brief to be detected by conventional nerve conduction techniques, but a summated volley of all the fibers within a nerve is large enough to be recorded, and it is this electrical potential and the one it produces in the muscle that it innervates that is utilized to study nerve function.
As nerve impulses pass centrifugally from the axon into its terminal branches, transmission may “break down,” especially if impulses arrive too frequently at branch points; they then fail to reach the neuromuscular junction of these fibers; or there may be failure of conduction at the neuromuscular junction, which is the characteristic feature of myasthenia gravis.
In large motor and sensory nerves, contiguous spread of action potentials along a fiber eventually decays over long distances. Conduction is aided by the structure and configuration of the myelin sheaths surrounding the axon. The sodium channels, which generate the action potential, are concentrated at short exposed segments of the axon, the nodes of Ranvier, lying between longer segments of myelinated axon, the internodes. In contrast, the internodes covered by myelin remain electrically insulated. This creates “flux lines” that converge on the nodes and allows current to be regenerated at each gap in the myelin. The speed of electrical conduction, which jumps in a “saltatory” fashion from node to node, is many times faster than conduction through an unmyelinated axon. The largest-diameter fibers have the thickest myelin sheaths and longest internodal distances, conferring on them the fastest conduction times.
Conventional laboratory studies of nerve conduction generally measure the speed of these fastest-conducting fibers. This dependence of nerve conduction on large, heavily myelinated fibers explains a number of electrophysiologic abnormalities that are a consequence of nerve disease. One common result, is a mild slowing of nerve conduction because of a dependence on the remaining smaller diameter, slower conducting axons. When myelin destruction is the prominent feature of a neuropathy (demyelinating neuropathy), conduction velocity is greatly slowed because of delays in regenerating the sodium current at nodes of Ranvier, or there may be total block of electrical conduction. An intermediate state of partial demyelination slows and desynchronizes the electrical volley, leading to temporal dispersion of the action potentials that reach the muscle. The cumulative effect of any of these changes is to reduce the number of nerve fibers that are capable of conducting an electrical volley, leading to a graduated reduction in the amplitude of the muscle action potential over longer segments of nerve. This reduction in amplitude of the compound motor action potential (CMAP) as the stimulating electrode is moved proximally is termed conduction block (discussed under “Studies of Nerve Conduction” further on). Blocked conduction of this restricted nature is a reliable marker of an acquired demyelinating neuropathy; of all the electrophysiologic changes, it also corresponds most closely to the degree of muscle weakness (see further on).
In contrast to these effects of demyelination of peripheral nerve, loss of axon fibers (axonal neuropathy) results in a reduction of the amplitude of summed electrical activity of the action potential in muscles uniformly all along the nerve, and to atrophic denervation of muscle as described further on.
The Neuromuscular Junction (Motor Endplate)
This is the interface between the finely branched nerve fiber and the muscle fiber, where the electrical activity of the motor nerve is translated into muscle action (Fig. 45-1). The nerve fiber contacts the muscle membrane in a trough-like junctional space of 50 mm—the synaptic cleft—between the axolemma and sarcolemma (see Fig. 49-1). Within the nerve terminal, a relatively fixed number of packets, or quanta, of acetylcholine (ACh), each packet containing about 10,000 molecules, are liberated through an exocytotic process that is triggered by the arrival of axonal action potentials. Arrival of the electrical impulse opens calcium channels in the presynaptic neural membrane, which serves to bind packets of ACh to the membrane and govern their release. Molecules of ACh diffuse into the synaptic cleft and attach to receptor sites on the postsynaptic membrane. Each impulse triggers the release of approximately 20 to 50 quanta of ACh and produces a depolarization of sufficient size to initiate an action potential in the postsynaptic muscle membrane through the same mechanism of a regenerative sodium current described earlier. Botulinum toxin interferes with exocytosis of ACh at the release site by cleaving “synaptic protein 25.” There is also a nonquantal release of ACh through continuous leakage. This appears to play a role in the trophic influence of nerve on muscle.
Figure 45-1.
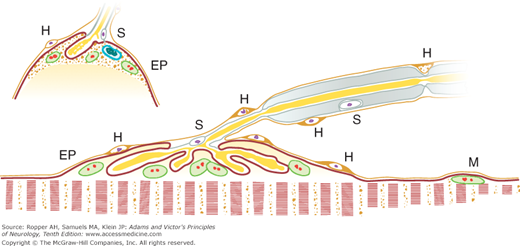
Motor endplate showing relationship between various structures in nerve and muscle. Last segment of myelin, with Schwann nucleus (S), terminates abruptly, leaving axis cylinder covered by sheaths of Schwann and Henle. Endplate nuclei (EP) of muscle fiber lie embedded in sarcoplasm and have same staining reactions as sarcolemmal nuclei (M). Ramifications of axis cylinder (telodendria) lie in grooves or pouches in granular sarcoplasm, each lined by spiny “subneural apparatus” of Couteaux, which is continuous with membranous sarcolemma and also Schwann membrane. Nucleus (S) of sheath of Schwann commonly lies near point of branching of axon. Sheath of Henle has small nuclei (H) and fuses with endomysial sheath of muscle fiber.
The ACh molecule binds to the postsynaptic ACh receptor, a complex of 5 proteins that constitute the postsynaptic ion channel. Binding of ACh by this receptor causes a conformational change in the receptor that leads to a local increase in the conductance of Na and K and other small ions. This produces a depolarization known as the endplate potential. Small (miniature) endplate potentials (MEPPs) are continuously formed and regenerated as the membranes repolarize, much as in the process of passive decay previously described. These potentials are too small to be recorded by routine electromyography (EMG) testing or to trigger an action potential, although fortuitous needle placement adjacent to a synapse may detect them.
The bound ACh is hydrolyzed by cholinesterase, a glycoprotein enzyme that exists in free form in the neuromuscular junction clefts. Its main function is to terminate the action potential and permit the sequential activation of muscle. The postsynaptic membrane, once depolarized, is refractory to another action potential until it is repolarized.
The calcium that entered the presynaptic nerve terminal is sequestered and then extruded, and the choline from the hydrolyzed ACh enters the nerve terminal, where it is resynthesized to ACh near the release sites.
The analysis of a rapid series of electrically of voluntarily elicited muscle contractions is used to test the function of the neuromuscular junction by stressing conduction across the junction. In general, a decrement in the amplitude of serial muscle action potentials is typical of postsynaptic failure, and an increment in the amplitude from a train of stimuli is a reflection of presynaptic failure.
Myasthenia gravis is the principal disease affecting the neuromuscular junction and represents a failure of postsynaptic function (see Chap. 49). In this process, the fundamental defect is not a deficiency of ACh or its release, but rather a failure of ACh to attach to the postsynaptic receptor, as a result of blockade by an antibody at the receptor site or of destruction of the membrane by antibody attack. There are several quite different synaptic disorders caused by botulism, aminoglycoside antibiotics, and the antibodies of the Lambert-Eaton myasthenic syndrome, which impede presynaptic release of ACh.
A number of pharmacologic agents also interfere with neuromuscular transmission by combining with the cholinergic (nicotinic) receptor on the postsynaptic membrane, thereby competitively blocking the transmitter action of ACh. The curariform drugs, derived from curare and termed nondepolarizing neuromuscular blockers because they do not alter the postsynaptic membrane potential are the main examples. Other drugs, notably succinylcholine and decamethonium, cause neuromuscular blockage by producing direct depolarization of the endplate and adjacent sarcoplasmic membrane (depolarizing neuromuscular blockers). Agents that inactivate cholinesterase have the opposite effect, i.e., they enhance the action of ACh. The ones in clinical use for the treatment of myasthenia gravis are the carbamates neostigmine, physostigmine, and pyridostigmine, the effects of which are reversible. The organophosphates are irreversible blockers of cholinesterase function, for which reason they are feared weapons of chemical warfare. Because the potent cholinergic antagonist atropine is active only at muscarinic sites, it has no effect at the neuromuscular junction.
Biochemistry of Muscle Contraction
The sarcolemma, the transverse tubules, and the sarcoplasmic reticulum each play a role in the control of the activity of muscle fibers. Figure 45-2 illustrates the structural components involved in excitation, contraction, and relaxation of muscle. Following nerve stimulation, an action potential is transmitted by the sarcolemma from the motor endplate region to both ends of the muscle fiber. Depolarization spreads quickly to the interior of the fiber along the walls of the transverse tubules, probably by a conducted action potential. The transverse tubules and the terminal cisternae of the sarcoplasmic reticulum come into close proximity at points referred to as triads. Here, depolarization of the transverse tubules alters the conformation of a voltage-sensitive calcium channel in the transverse tubule membrane. This causes a large protein (the ryanodine receptor) in the membrane of the adjacent sarcoplasmic reticulum (SR) to open an internal calcium pore, allowing stored calcium to exit the SR and flood into the cytoplasm. The released calcium binds to the regulatory protein troponin, thereby removing the inhibition exerted by the troponin–tropomyosin system upon the contractile protein actin. This allows an interaction to occur between the actin molecules of the thin filaments and the cross-bridges of the myosin molecules in the thick filaments and enables myosin ATPase to split adenosine triphosphate (ATP) at a rapid rate, thereby providing the energy for contraction. This chemical change allows the filaments to slide past each other, thereby shortening the muscle fiber. Relaxation occurs as a result of active (energy-dependent) Ca reuptake by the sarcoplasmic reticulum.
Figure 45-2.
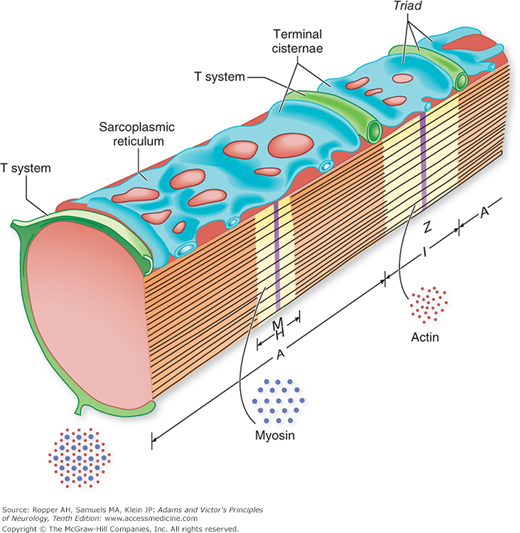
Schematic of the major subcellular components of a myofibril. The transverse (T) system, which is an invagination of the plasma membrane of the cell, surrounds the myofibril midway between the Z lines and the center of the A bands; the T system is approximated to, but apparently not continuous with, dilated elements (terminal cisternae) of the sarcoplasmic reticulum on either side. Thus, each sarcomere (the repeating Z-line-to-Z-line unit) contains two “triads,” each composed of a pair of terminal cisternae on each side of the T tubule. (From Peter, by permission.)
The pyrophosphate bonds of ATP, which supply the energy for muscle contraction, must be replenished constantly by a reaction that involves interchanges with the muscle phosphagen creatine diphosphate, where high-energy phosphate bonds are stored. These interactions, in both contraction and relaxation, require the action of creatine kinase (CK).
Myoglobin, another important muscle protein, functions in the transfer of oxygen, and a series of oxidative enzymes are involved in this exchange. The intracellular Ca, as noted earlier, is released by the muscle action potential and must be reaccumulated within the cisternae of the sarcoplasmic reticulum before actin and myosin filaments can slide back past one another in relaxation. This reuptake of Ca requires the expenditure of considerable energy. When there is defective generation of ATP, for example, from an enzyme deficiency, the muscle remains shortened, as in the contracture of phosphorylase deficiency (McArdle disease) or of phosphofructose kinase deficiency. The same sort of shortening contracture occurs under normal conditions in some of the “catch muscles” of certain mollusks and is the basis of rigor mortis in mammals.
Many glycolytic and other enzymes (transaminases, aldolase, CK) are utilized in the metabolic activity of muscle, particularly under relatively anaerobic conditions. Muscle fibers differ from one another in their relative content of oxidative and glycolytic enzymes; the latter determine the capacity of the muscle fiber to sustain anaerobic metabolism during periods of contraction with inadequate blood flow. Muscle cells rich in oxidative enzymes (type 1 fibers) contain more mitochondria and larger amounts of myoglobin (therefore appearing red), have slower rates of contraction and relaxation, fire more tonically, and are less fatigable than muscle fibers poor in oxidative enzymes. The latter (type 2 fibers) fire in bursts and are used in quick phasic, rather than sustained, reactions. The amount of myosin ATPase activity, which governs the speed of contraction, is low in oxidative-rich fibers and high in glycolytic-rich fibers. The myosin ATPase stain at pH 9.4 has been used to differentiate these two types of fibers in microscopic sections. Type 1 fibers have a low content of myosin ATPase, and type 2 (phosphorylative-rich) fibers have a high content of this enzyme; hence type 1 fibers stain lightly and type 2 darkly (the reverse reaction occurs at pH 4.6). All the fibers within one motor unit are of the same type, a feature that is used to advantage to identify the reinnervation of muscle fibers by a single motor neuron after adjacent neurons have died and denervated their constituent muscle fibers (fiber-type grouping).
The chemical energy required to maintain the various activities of the muscle cell is derived mainly from the metabolism of carbohydrate (blood glucose, muscle glycogen) and from fatty acids (plasma-free fatty acids, esterified fatty acids, and ketone bodies). There is a lesser contribution to energy from branched-chain and other amino acids, but their activity increases during prolonged exercise.
The most readily available source of muscular energy is glycogen, which is synthesized and stored in muscle cells. It provides more than 90 percent of the energy needs of muscle under conditions of high work intensity and during the early stages of submaximal exercise. Blood glucose and free fatty acids supplement intracellular glycogen as exercise proceeds. The free fatty acids are obtained from endogenous triglycerides (found mostly in type 1 fibers), from the triglycerides released by circulating lipoproteins, and from the lipolysis of adipose tissue. Most of the energy needs of resting muscle are provided by fatty acids.
The enzymatic reactions involved in the transport of these substrates into muscle cells and their intracellular synthesis and degradation during anaerobic and aerobic cell conditions have been thoroughly investigated and most of the participating enzymes have been identified. This subject is too extensive to be presented in a textbook of neurology, but enough is known about these matters to state that there are diseases that impair the contractile functions of muscle without destroying the fiber. In particular, specific enzymatic deficiencies alter carbohydrate utilization (myophosphorylase, debrancher enzyme, phosphofructokinase, and phosphoglyceromutase deficiencies), fatty acid utilization (carnitine and carnitine palmitoyl transferase deficiencies), pyruvate metabolism, and cytochrome oxidase activity (in the mitochondrial muscle diseases). These diseases are discussed in later chapters.
Physiology of Muscle Contraction
The contraction of a muscle fiber may be viewed as a series of electrochemical events that culminate in a mechanical effect. The mechanical change far outlasts the electrical one and extends through the period when the muscle fiber is refractory to another action potential. When a second muscle action potential arrives after the refractory phase of the previous action potential, but before the muscle has relaxed, the contraction will be prolonged. Thus, at frequencies of anterior horn cell firing of more than 100 per second, twitches fuse into a sustained voluntary contraction or fused tetanus. In most sustained contractions, there is incomplete tetanus, attained by firing rates of 40 to 50 per second. In this fashion, the mechanical contractions of individual muscle fibers are smoothed into a continuous process, even though the electrical potentials present as a series of depolarizations, separated by intervals during which the muscle membrane resumes its resting polarized state.
As pointed out in Chap. 3, the physiology of muscle activity is best considered in terms of motor units, i.e., the group of muscle fibers innervated by a single anterior horn cell. The strength of muscle contraction is in turn a function of the number and rates of firing of many adjacent motor units. The smoothness of contraction depends on the integrated and sequential enlistment of motor units of increasing size. The electrical signal of this summated contraction, the compound motor action potential, or CMAP, as recorded at the skin surface overlying a muscle is the main feature of the surface EMG and serves as the basis for quantifying the size of the motor nerve potential in the nerve conduction examinations.
The CMAP can be visualized on the screen of an oscilloscope or a computer and its onset used to measure the speed of motor nerve conduction (conduction velocity) as well as the summated amplitudes produced by all the innervating nerve fibers. It can also be converted into an audible noise. Further study involves the insertion into the muscle of a coaxial needle, which samples several motor units in the vicinity of the electrode. When elicited by a sustained voluntary contraction, the flurry of electrical activity from many muscle fibers at different distances from the electrodes is referred to as an interference pattern (see further on, under “Studies of Nerve Conduction” and “Needle Examination of Muscle [Electromyography]”).
Various biochemical changes may cause not only an impairment of muscular activity (paresis, paralysis) but also excessive irritability, tetany, spasm, and cramp. In these instances, spontaneous discharges may occur from instability of axon polarization; hence a single nerve impulse may initiate a train of action potentials in nerve and muscle, as in the tetany of hypocalcemia. The common cramps of calf and foot muscles (painful, sustained contractions with motor unit discharges at frequencies up to 200 per second) may be a result of increased excitability (or unstable polarization) of the motor axons. Quinine, procainamide, diphenhydramine, and warmth reduce the irritability of nerve and muscle fiber membranes, as do a number of antiepileptic drugs that act by blocking sodium channels, thereby limiting spontaneous membrane discharges.
The muscle fiber, which is wholly dependent on the nerve for its stimulus to contract, may be physiologically activated or paralyzed in a number of ways that are explained by the principles described above. The main disturbances of nerve, muscle, and neuromuscular function occur when the motor nerve cell or its axon is injured or inexcitable, in which case the muscle cannot be stimulated; or, the nerve cell in the anterior horn of the spinal cord may be disinhibited, permitting the discharge of continuous action potentials, as in tetanus and the “stiff man” syndrome (see Chap. 48); the nerve fiber may fail to conduct impulses (demyelinating neuropathies) or the number of fibers may be inadequate to produce a fused and sustained contraction (axonal neuropathies); the distal axon may not distribute the nerve impulse simultaneously to all parts of the motor unit; ACh may not be released at the presynaptic region of the neuromuscular junction (as occurs in botulism and in the Eaton-Lambert syndrome) or, once released, ACh may not be inactivated by cholinesterase (physostigmine, organophosphates); the receptor zone on the postsynaptic membrane may be destroyed or blocked by antibodies or pharmacologic agents (myasthenia gravis or curariform drugs); and, finally, the metabolic or contractile elements of the muscle may not react or, once contracted, may not relax. One type of this last category is due to genetically determined abnormalities of voltage-gated ion channels, the “channelopathies,” that are the subject of Chap. 50.
Similarly, there may be an unstable polarization of the nerve fibers, as in tetany and in dehydration with salt depletion, or hyperirritability of the motor unit, as in amyotrophic lateral sclerosis. The threshold of mechanical activation or electrical reactivation of the sarcolemmal membrane may be reduced, as occurs in myotonia, or impairment of an energy mechanism within the fiber may slow the contractile process, as in hypothyroidism; or a deficiency of phosphorylase, which deprives muscle of its carbohydrate energy source, may prevent relaxation, as in the contracture of McArdle disease. Lesions of the most peripheral branches of nerves, which allow nerve regeneration, may give rise to continuous activity of motor units. This is expressed clinically as a rippling of muscle, or myokymia.
In recent years, special techniques have made it possible to study each of the proteins and channels involved in neuromuscular transmission and the excitation-contraction-relaxation of muscle fibers. The amino acid composition of these proteins has also been determined. This information is being increasingly applied to the analysis of genetic diseases of muscle in the normal state and under conditions of disease. Pertinent comments and references to this subject are found in the chapters that follow.
Effects on Muscle of Serum Electrolytes
Diffuse muscle weakness or muscle twitching, spasms, and cramps should always raise the question of uremia or an abnormality in serum electrolytes. These disorders reflect the concentrations of electrolytes in the intra- and extracellular fluids. If the plasma concentration of K falls below 2.5 mEq/L or rises above 7 mEq/L, weakness of extremity and trunk muscles results; below 2 mEq/L or above 9 mEq/L, there is almost always flaccid paralysis of these muscles and later, of the respiratory ones as well; only the extraocular and other cranial muscles are spared. In addition, the tendon reflexes are diminished or absent. The normal reaction of muscle to direct percussion is also reduced or abolished, suggesting impairment of transmission along the sarcolemmal membranes themselves. Hypocalcemia of 7 mg/dL or less (as in rickets or hypoparathyroidism) or relative reduction in the proportion of ionized calcium (as in hyperventilation) causes increased muscle irritability and spontaneous discharge of sensory and motor nerve fibers (i.e., tetany) and sometimes convulsions from similar effects upon cerebral neurons; frequent repetitive and prolonged spontaneous discharges grouped in couplets or triplets appear in the EMG. Hypercalcemia with Ca levels above 12 mg/dL (as occurs in vitamin D intoxication, hyperparathyroidism, carcinomatosis, sarcoidosis, and multiple myeloma) causes weakness perhaps on a central basis, and lethargy. Extreme hypophosphatemia, observed most often with intravenous hyperalimentation or bone tumor, can cause acute areflexic paralysis with nerve conduction abnormalities. Reduction in the plasma concentration of magnesium also results in tremor, muscle weakness, tetanic muscle spasms, and convulsions; a considerable increase in magnesium levels leads to muscle weakness and depression of central nervous function. The weakness of muscle in hypermagnesemia may also be partly a result of reduced release of ACh at the motor endplate.
Changes in Serum Levels of Enzymes Originating in Muscle Cells
In all diseases that cause extensive damage to striated muscle fibers, intracellular enzymes leak out of the fibers and enter the blood. Those measured routinely are the transaminases, lactic acid dehydrogenase, aldolase, and, especially, CK. The concentration of CK in serum has proved to be the most consistent and most sensitive measure of skeletal muscle damage. Because high concentrations of this enzyme are found in heart muscle and brain, raised serum values may be a result of myocardial or cerebral infarction, as well as of the necrotizing diseases of striated muscle (polymyositis, muscle trauma, muscle infarction, and the more rapidly advancing muscular dystrophies). For serum CK levels to be interpretable, one has to be certain that the enzyme released into the serum is not derived from heart or brain. This can be determined by the quantitation of serum isoenzymes of CK, referred to as MB, MM, and BB (M, muscle; B, brain); their measurement provides a means for the detection of damage to myocardium (MB), skeletal muscle (MM), and nervous tissue (BB), respectively.
The MM form of CK is found in highest concentration in striated muscle, but these muscles also contain 5 to 6 percent MB isoform. Myocardial tissue contains 17 to 59 percent MB; hence, the diagnosis of myocardial infarction requires that the CK-MB fraction be greater than 6 percent (or that troponin, which is overrepresented in heart muscle, be elevated in the serum). Embryonic and regenerating muscle contains more CK-MB than mature normal muscle. In patients with destructive lesions of striated muscle, serum values of CK often exceed 1,000 U/L and may reach 50,000 U or more (the upper limit of normal varies from 65 to 300 U/L, depending on the method of measurement). It is notable that in some black men the level may normally be in the range of 500 U/L in the absence of muscle or nerve disease (see below). The serum of the healthy adult contains only the MM isoenzyme, but in healthy children, as much as 25 percent of serum CK may be derived from the MB fraction.
As interesting is the rise in serum enzyme concentration in some children with progressive muscular dystrophy before there is enough destruction of fibers for the disease to be clinically manifest, at least as judged by the relatively crude test of muscle strength. Moreover, the unaffected female carrier of Duchenne dystrophy may be identified by an elevated serum level of CK; these changes reflect a “leaky” membrane that allows the transgression of the large enzyme molecules. Alterations of serum enzyme levels are, however, nonspecific as they occur in all types of disease that damage the muscle fiber. Moreover, in the more slowly evolving types of dystrophy, the serum levels of CK may be normal.
It would be expected that enzyme values would be normal in paralysis due to denervation with secondary muscular atrophy, but elevations are sometimes observed in patients with progressive spinal muscular atrophy and amyotrophic lateral sclerosis, particularly if there is relatively rapid progression of the disease. Even vigorous exercise or surgical operations involving muscle elevate CK, sometimes with the MB fraction exceeding 6 percent.
In some normal individuals, CK may be persistently elevated without evidence of muscle or other disease. There is a particularly high incidence of this finding, albeit of mild degree, in African American men. An unexplained alteration of the sarcolemma with elevated serum CK occurs in hypothyroidism and in alcoholism. Toxic myopathies, for example, the type caused by the cholesterol-lowering drugs (statins), are another common cause of elevation in CK in current practice. Among the causes of persistently elevated CK are numerous underlying genetically determined muscle defects, usually one of the varieties of abnormal dystrophin or another membrane protein. Detailed examination may reveal slight proximal weakness and perhaps minimal calf enlargement characteristic of the mildest forms of Becker dystrophy. Closely related is the finding of CK elevations that occur in females who are asymptomatic carriers of Duchenne or Becker dystrophies. These diseases are described in Chap. 48.
Several nondystrophic muscle diseases also present as elevated CK levels in the blood, including polymyositis and dermatomyositis, carnitine palmityl transferase deficiency, some of the mitochondrial myopathies, McArdle disease, central core disease, multicore disease, and some forms of inclusion body myopathy. Finally, the rapid denervation of muscle that accompanies some cases of ALS may cause CK elevation.
If other sources of persistently elevated CK levels have been excluded, particularly those caused by exercise, trauma, and by drug-induced muscle damage, it is appropriate to follow the patient over time so that mild weakness may be detected, or to perform a biopsy to resolve the issue of an inflammatory myopathy or a mild muscular dystrophy. Because treatment would likely be withheld until weakness or pain arises, in idiopathic cases of raised enzyme levels, it seems prudent to delay biopsy unless the enzyme levels are massively elevated.
An isolated elevation of aldolase, the serum enzyme other than CK that is derived predominantly from skeletal muscle, generally has less clinical significance. Measurement in the serum of various transaminases or lactate dehydrogenase is not particularly useful for the diagnosis of muscle disease because of the ubiquitous distribution of these enzymes in many mammalian tissues. Nevertheless, the neurologist should be aware that unexplained elevations in all of the muscle-derived enzymes (CK, lactate dehydrogenase [LDH], serum glutamic-oxaloacetic transaminase [SGOT], aldolase) can be caused by inevident muscle trauma and by many other processes that damage the muscle membrane. As mentioned, cardiac muscle-specific enzyme, troponin, is not expressed in skeletal muscle and therefore is not found in the serum in cases of muscle disease, except in unusual circumstances in which regenerating muscle fibers in muscular dystrophies may transiently express the enzyme (the T, but the I, isotype) as noted in the series reported by Jaffe and colleagues. More often, elevations or troponin in these muscular diseases reflects a parallel myocardial disorder that accompanies several varieties of dystrophy.
Myoglobinuria
The red pigment myoglobin, responsible for much of the color of muscle, is an iron-protein compound present in the sarcoplasm of striated skeletal and cardiac fibers. Of the hematin compounds, approximately 25 percent are in muscle and the remainder are in red blood corpuscles and other cells. Destruction of striated muscle, regardless of the cause liberates myoglobin, and because of its relatively small size, the molecule filters through the glomeruli and appears in the urine, imparting to it a burgundy-red color. Because of the low renal threshold for myoglobin, excretion of the pigment is so rapid that the serum remains uncolored. In contrast, the high renal threshold for hemoglobin colors both the serum and the urine if there is destruction of red blood corpuscles. Myoglobinuria should thus be suspected when the urine is deep red and the serum is normal in color. It is estimated that 200 g of muscle must be destroyed to color the urine visibly (Rowland).
As with hemoglobinuria, the guaiac and benzidine tests performed on urine are positive if myoglobin is present. However, the colored urine does not fluoresce, as it does in porphyria. On spectroscopic analysis, myoglobin shows an absorption band at 581 nm but the most sensitive method for measuring myoglobin in the urine and serum is by radioimmunoassay. Some years ago, the measurement of creatine and creatinine in blood and urine was a standard method of estimating damage to striated muscle. This technique is now seldom used, having been replaced by the measurement of CK and its isoenzymes. However, measurement of urine creatinine in large-volume samples of urine that have been collected for other testing assures that an adequate amount of renal excretion is being captured in the sample.
Related posts:
 Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
 Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
 Chapter 48. Diseases of Muscle
Chapter 48. Diseases of Muscle
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


