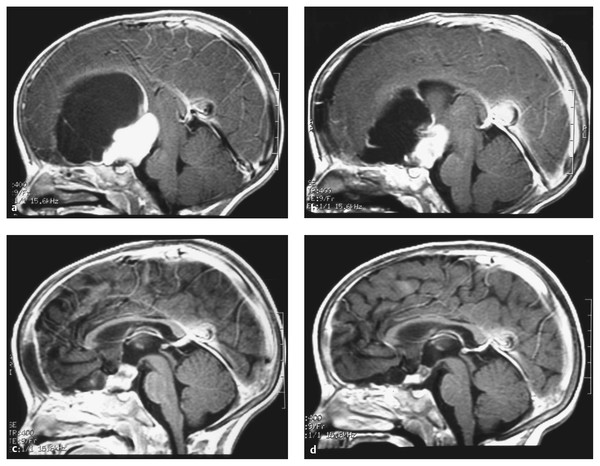
The current management of primary central nervous system (CNS) tumors of childhood has resulted in slowly improving survival rates and has become increasingly complex. Until the late 1970s and early 1980s, treatment for children with both benign and malignant brain tumors primarily consisted of surgery with or without radiation therapy. Chemotherapy was restricted to children with recurrent malignant brain tumors, usually with the goal of transiently prolonging life. A series of clinical investigations completed over the past quarter century has demonstrated the efficacy of chemotherapy for children with recurrent and newly diagnosed malignant and benign primary CNS tumors. For some brain tumors, such as medulloblastomas and possibly high-grade gliomas, chemotherapy at the time of diagnosis, when added to radiation therapy, improves the likelihood of survival.1–3 Furthermore, for some children with nondisseminated disease, chemotherapy may allow a reduction in the dose of radiotherapy needed.4 In infants and very young children, chemotherapy is being increasingly used to delay, if not obviate, the need for alternative treatments.5 To date, the chemotherapies used to treat brain tumors have been primarily conventional cytotoxic agents, which relatively nonselectively kill tumor cells. However, recently, a host of biological agents have become available that are designed to target tumor cells and are in clinical trials.6,7 The specific role of this or any other type of chemotherapy in the management of childhood brain tumors is still being defined. Discussions of the role of chemotherapy for childhood brain tumors have to take into account many factors. Childhood primary tumors of the CNS comprise a heterogeneous group of lesions. Generalizations are difficult to make, given the histologic and biological heterogeneity of these lesions.8,9 Even within recognized groupings—for example, medulloblastomas—there are now well-accepted molecular subgroupings, with likely different sensitivities to treatment and different prognoses.10 Obviously, for a chemotherapeutic agent to be effective, the tumor must be sensitive to the agent employed. The selection of a drug or drug regimen that is most likely to be effective for a given type of tumor has been primarily based on clinical studies investigating the response of the tumor to treatment.11 Laboratory methods have been established to facilitate analysis of the biology and the therapeutic profile of a given tumor. Such laboratory approaches result in a more rational choice of chemotherapeutic agents to be evaluated. Agents with possible activity are tested by in vitro methods, and selected agents are further studied in animal models, such as a xenograft model in which human tumors are grown in immunodeficient (nude) mice. This rigorous means of testing allows potentially active new compounds to be identified before clinical trials are undertaken. For biological agents, which may affect not only specific targets, such preclinical testing is likely even more important. Yet other factors, such as the ability of an agent to reach a tumor in effective concentrations, remain crucial. The majority of drugs presently in use were chosen on the basis of their efficacy against other forms of cancer and subsequent results in clinical trials in adults and children with brain tumors (▶ Table 50.1). In general, these clinical trials can be separated into three major categories (▶ Table 50.2). Phase I trials are the initial trials, and the purpose of such studies is to determine the maximum tolerated dose of the drug, to identify its toxicities, and often to perform detailed pharmacokinetic studies. These studies are carried out in children with recurrent disease in whom all proven means of therapy have been exhausted. In pediatrics, the majority of drugs used for patients with brain tumors in Phase I testing have already been demonstrated to be somewhat efficacious in adults with primary CNS tumors. Many of the active antitumor agents now in use for patients with newly diagnosed brain tumors initially were tested in these Phase I clinical trial settings. Once the optimal dose and schedule of a drug have been defined in a Phase I study, a Phase II trial is undertaken to determine the response rate of a specific tumor to the drug. The predominant measure of the efficacy of a drug in such trials is the objective response of the tumor to treatment, with a more than 50% reduction in tumor size on neuroradiographic studies considered a partial response and a complete disappearance of tumor considered a complete response. However, time to progression may also be used as a measure of effectiveness. Phase II trials are predominantly undertaken in children with recurrent disease. However, for some tumor types, especially those that are highly resistant to any known form of treatment, preradiation chemotherapeutic Phase II trials (which have also been titled neoadjuvant trials) have been performed. Reasons for using preradiation chemotherapeutic trials include that patients may be better able to tolerate a drug or drugs before other forms of therapy (primarily radiation therapy) have been delivered and that prior therapy may make a tumor more resistant to the effects of chemotherapy, thus causing active agents to be incorrectly considered ineffective. The final step in the evaluation of the efficacy of a chemotherapeutic agent or a group of drugs is a randomized comparison of the efficacy of the drug or regimen, either in addition to conventional therapy or instead of conventional therapy, versus that of standard therapy. Given the relative rarity of pediatric CNS tumors, Phase III studies usually must be undertaken in a multi-institutional setting. The efficacy of a chemotherapeutic agent is not dependent solely on the sensitivity of a tumor to the drug. Other factors are also of importance, including the following: pharmacokinetics of the drug, host pharmacogenomics, concurrent use of medications that may alter drug metabolism through the cytochrome P-450 system of the liver, ability of the chemotherapeutic agent to reach the tumor site in high enough concentrations to affect tumor growth, ability of the tumor cells to accumulate and retain the drug, and ability of the tumor cells to repair drug-induced damage.12–14 A great deal of emphasis has been placed on the delivery of the drug, or drugs, to the tumor, but increasing information suggests that equally important factors may be the cellular mechanisms within the tumor responsible for drug resistance. The blood–brain barrier has been identified as a major reason for the ineffectiveness of certain chemotherapeutic agents.15–17 In some pediatric brain tumors, such as medulloblastomas, which often have an abundant blood supply, the significance of the blood–brain barrier is unclear. For infiltrating gliomas, it is well documented that drug delivery is a more important issue because it is often difficult to deliver adequate concentrations of drugs to the periphery of the tumor.15–17 A variety of means have been used in attempts to improve drug delivery, including higher doses of chemotherapeutic agents, osmotic agents to disrupt the barrier, and more recently chemotherapeutic agents coupled with other drugs that selectively disrupt the tumor blood–brain barrier.15–19 For example, the anti-angiogenesis drug cilengitide, which targets integrins on neoendothelium, was found to improve the outcome of adult patients with glioblastoma multiforme (GBM) when it was administered with temozolomide chemotherapy; however, improvement was demonstrated only in those patients with tumors exhibiting MGMT (O6-methylguanine DNA-methyltransferase) promoter methylation, which confers sensitivity to temozolomide. This finding suggests that in this setting cilengitide was helping to increase the effective concentration of temozolomide delivered to temozolomide-sensitive tumors through its effect on normalization of the tumor vasculature.20 The goal of high-dose chemotherapy is to increase the delivery of cytotoxic agents to tumor effectively by overcoming the limited permeability of the blood–brain barrier. Predominantly lipid-soluble alkylating agents with nonoverlapping hematologic toxicities have been chosen. Initially, trials were performed with one cycle of chemotherapy supported by autologous bone marrow transplant, but this type of support has been primarily supplanted by peripheral blood stem cell support. Toxicity with autologous bone marrow transplant protocols has been significant; in early trials, it was associated with a 5 to 15% treatment-related mortality rate.21–23 In recent investigations, the administration of less intensive drug regimens multiple times with peripheral blood stem cell support has resulted in decreased morbidity and mortality.24 Other methods, such as intra-arterial therapy, intracavitary therapy, the use of biodegradable polymers intraoperatively impregnated with a chemotherapeutic agent (allowing a slow local release of the agent—wafer therapy), and direct intraparenchymal infusional therapy, are also being used in attempts to increase delivery to the local tumor site.25–31 Most recently, the use of circulating nanoparticles conjugated to tumor-targeting molecules and radioisotopes, chemotherapeutic agents, or other biological agents has been explored experimentally with some success.32,33 The first human adult cancer trials using therapeutic nanoparticles started in 2012. Direct delivery to tumors via the infusion of toxins conjugated to agents that selectively bind tumor cells is under study in both adults and children with brain tumors. Such approaches, which use convection delivery techniques, are appealing yet potentially extremely toxic. Convection delivery has been shown to deliver agents quite extensively throughout the brain, but it is unclear whether the conjugated toxin is best delivered into the tumor or in the area around the tumor. The selectivity of tissue damage is also unclear. Although these are potentially useful techniques, their efficacy has not yet been clearly shown in either adults or children with brain tumors. The mechanisms of drug resistance in primary CNS tumors are actively being defined.34 Resistance to a given therapy may be an intrinsic property of a tumor or may be acquired after treatment. Tumors may acquire resistance by the loss of normal properties or by the amplification of existing mechanisms in the cells. Tumors may become cross-resistant to a whole group of drugs by a similar mechanism. Mechanisms of drug resistance include alterations in cell transport, the expression or amplification of genes like the multidrug resistance gene, and elevated intracellular levels of enzymes that interfere with the antitumor activity of a drug. The efficacy of the alkylator agents, at present one of the most widely used classes of drugs for the treatment of CNS tumors, is decreased by elevated levels of intracellular aldehyde dehydrogenase and glutathione or glutathione S-transferase.35–38 Similarly, intrinsic or acquired resistance to the nitrosoureas may be due to increased repair of DNA mediated by the O6-alkyl guanine DNA alkyl transferase protein or by elevated levels of thiol or glutathione S-transferases.35–38 A variety of techniques have been used in an attempt to overcome mechanisms of drug resistance, such as saturation of enzymes by alternative substrates or inactivation of cell resistance mechanisms by drugs, but none have shown a clear improvement in efficacy. One such approach under active study is the drug O6-benzylguanine, which depletes alkyl glutathione S-transferase levels. This enzyme-depleting drug was initially given with bischlorethyl-nitrosourea (BCNU) but is now also under study to increase sensitivity to temozolomide, as well as to improve the efficacy of alkylator-impregnated polymer wafers placed, after tumor resection, into the rim of the resection cavity. In 1979, Goldie and Coldman proposed a mathematical model that related curability of a malignancy to the appearance of resistant cell lines.39,40 They proposed that control of a neoplasm was a function of various factors favoring resistance, including the tumor’s spontaneous mutation rate. An outgrowth of this theory is the concept that a tumor is less likely to be resistant to multiple agents administered simultaneously than it is to be resistant to individual agents because cells are not given an opportunity to mutate and develop pleiotropic multiple-drug resistance. Additionally, if drugs are given over a relatively short time, myelosuppression should be less because damage to hematopoietic precursor cells depends partially on the duration of exposure. This concept has been widely tested in pediatric brain tumor trials. The majority of ongoing chemotherapeutic studies are using a combination of drugs rather than single agents in attempts to improve efficacy. In pediatrics, the applications of chemotherapy have not been limited to the development of means to improve survival. Given the well-documented toxicities of radiation, especially whole-brain irradiation, chemotherapy has also been employed in attempts to reduce the amount of radiotherapy required for disease control and in some cases to delay, if not obviate, the need for radiotherapy.4,5 Chemotherapy is now widely used for infants and young children with malignant brain tumors and for selected patients with low-grade tumors that are not surgically resectable without prohibitive morbidity. Conventional chemotherapy has consisted predominantly of drugs that cause cytotoxic damage, with considerable systemic and, at times, CNS toxicity. In theory, the use of biological therapy, which in principle selectively targets specific molecular aspects of the tumor, results in a better therapeutic window and the ability to deliver therapy with less toxicity and greater efficacy. This concept of sparing normal cells by using molecularly based therapy is intriguing and in many ways is the “Holy Grail” of future cancer treatment. However, currently available biological agents may be less specific in the targets they hit than originally thought, and the molecular mechanisms disrupted may also be critical for normal cellular function in the brain or other organs. Concepts of cellular signaling, signal transduction pathways involved in cell regulation, proliferation, and survival are rapidly evolving.41–43 The role of proto-oncogenes and tumor suppressor genes in some brain tumors is just beginning to be characterized. Other factors involved in cancer development and growth, such as neoplastic angiogenesis and tumor invasion and migration, are also likely crucial in sustaining tumor growth and facilitating tumor spread. Agents have recently been developed that target critical mechanisms for tumor growth and are quickly entering the therapeutic arena. However, to date, none of them have demonstrated clear efficacy in regard to long-term control of pediatric brain tumors, and their short- and long-term toxicities are still being elucidated. At the same time, because of the unique mechanisms of action and promise of these agents, there is tremendous interest in integrating them, as rapidly as possible, into the standard treatment of childhood brain tumors. Receptor tyrosine kinases are transmembrane receptor proteins that regulate many critical cellular processes, including proliferation and cell survival.43–45 A host of tyrosine protein kinase inhibitors have been developed in attempts to arrest growth signaling. In tumors, signaling may be activated by a variety of different mechanisms, including overexpression of the tyrosine kinase and gain of function; at times, the receptor is constitutively activated (especially in adult cases of GBM) even without the presence of a ligand.45 A variety of receptor tyrosine kinases have been implicated in brain tumor pathogenesis and thus are potential targets for therapy. These include platelet-derived growth factor, epidermal growth factor, vascular endothelial growth factor, insulin-like growth factor, and fibroblast growth factor.6,46 The biological agents that have been used include small-molecule inhibitors, which target the intercellular tyrosine kinase domains, and monoclonal antibodies, which act predominantly against growth factor ligands. Platelet-derived growth factor receptor, which predominantly comprises two receptors (alpha and beta), has been shown to mediate cell signaling pathways involved in tumor proliferation, survival, invasion, and angiogenesis. Amplifications of platelet-derived growth factor have been demonstrated in pediatric high-grade gliomas, diffuse intrinsic brainstem gliomas, and medulloblastomas.47,48 Imatinib, a small-molecule inhibitor of BRC/ABL kinase, KIT, and platelet-derived growth factor receptor, has been found to be useful in a variety of different tumor types; however, studies of brain tumors in children have found it to be relatively ineffective,49,50 possibly because of its poor CNS penetrance. Other platelet-derived growth factor receptor inhibitors are presently in clinical trials. Epidermal growth factor receptor comprises four members and is expressed on actively dividing cells, not only in brain tumors but also in brain neurogentic niches, such as the subventricular zone.43,44,51–53 Overexpression and amplification of epidermal growth factor receptor have been reported in pediatric high-grade gliomas and brain stem gliomas, as well as ependymomas, medulloblastomas, and low-grade gliomas.50–53 A variety of monoclonal antibody drugs have been in clinical trials, with small molecules used to target this family of receptors.54–56 Although these drugs have had a reasonable toxicity profile, their overall efficacy in regard to either tumor response or prolongation of survival has yet to be proved in pediatrics. Small-molecule inhibitors such as erlotinib, gefitinib, and lapatinib have also been fairly extensively tested against different types of pediatric brain tumors with some hints of efficacy, especially in specific subsets of patients, but no clear benefit in any specific tumor type. Vascular endothelial growth factor, which comprises three receptors, has been identified as a potential target means to control tumor growth.57 Vascular endothelial growth factor and vascular endothelial growth factor receptor increase with hypoxia, thus contributing to further angiogenesis, and act as another means to enhance tumor growth (oxygen tension is lower in rapidly progressing tumors than in normal tissue environments).58 Monoclonal antibodies and small-molecule inhibitors of vascular endothelial growth factor ligands and receptors are in active development. Bevacizumab, a monoclonal antibody against vascular endothelial growth factor, has been approved for the treatment of recurrent malignant gliomas in adults.59 Multiple studies have been undertaken in pediatrics without clear benefit in trials of adult malignant glioma or brain stem glioma.60 There is evidence of efficacy of bevacizumab used in combination with irinotecan for pediatric low-grade gliomas.61 Some anti-angiogentic drugs have caused significant toxicities, such as difficult-to-control hypertension and proteinuria. A major risk associated with vascular endothelial growth factor receptor drugs, as well as possibly other growth factor receptor inhibitors affecting vasculature, has been intratumoral hemorrhage. To date, this risk has not been rate-limiting in pediatrics but remains significant nonetheless. Downstream signal transduction pathways have also been targeted in pediatric brain tumors, especially the RAS/mitogen-activated protein kinase pathway, which plays an important role in tumor cell proliferation and survival.62 This RAS/RAF/MEK/ERK pathway offers a multitude of targets.62 Farnesyl transferase inhibitors, which inhibit the post-translational modification of RAS, have been studied but have not been found to be particularly effective.63,64 This may be because they are not selective as to the type of RAS abnormality in the tumor or do not adequately penetrate the tumor. RAF inhibitors, such as sorafenib, are in clinical trials.65 Many RAF inhibitors actually are really multi-kinase inhibitors with activity against a variety of signal transduction targets. Although initially it was believed that drugs with great specificity would be most effective, there is a school of thought in developmental therapeutics that agents targeting multiple pathways, so-called multi-kinase inhibitors or “dirty kinase” inhibitors, may be even more effective. Sunitinib, like sorafenib, is a multi-kinase inhibitor that is in clinical trials.66 The MEK inhibitors are still another class of drugs that are being actively evaluated. Because of the findings of BRAF oncogene activation in a high proportion of grade I (pilocytic) astrocytomas, there is significant interest in using such inhibitors in pediatrics.67,68 The phosphatidylinositol 3-kinase/protein kinase/mammalian target of rapamycin (mTOR) pathway is still another pathway being actively targeted in pediatric brain tumors.69,70 There has been enthusiasm concerning the use of rapamycin since initial experience in patients with tuberous sclerosis who harbored giant cell astrocytomas demonstrated an excellent response rate to rapamycin or a similar mTOR inhibitor drug, RAD001.71 How well this type of activity can be generalized to other brain tumors is under active study. Drugs specifically targeting P13 kinase activity are early in study, as are mTOR/P13k dual inhibitors. The sonic hedgehog pathway seems to play a significant role in the pathogenesis of subsets of medulloblastomas. Sonic hedgehog inhibitors are actively being studied in this tumor type, and there is interest in potentially evaluating the efficacy of these drugs in other CNS tumors.72 Similarly, the Notch signaling pathway, which has demonstrated an important role in maintaining neural progenitor cells while inhibiting differentiation, has been noted to be dysregulated in gliomas, medulloblastomas, and ependymomas.73,74 γ-Secretase is one of the main proteolytic enzymes that cleaves the receptor and activates the Notch cascade. γ-Secretase inhibitors are a means to decrease Notch signaling. Histone deacetylase inhibitors are being actively explored in pediatric brain tumors.75 Histone deacetylase is involved in chromatin condensation and epigenetic silencing. Interestingly, one of the more commonly used anticonvulsants, valproic acid, is a histone deacetylase inhibitor.76–78 Valproic acid and other drugs, such vorinostat and desipeptide, are in clinical trials. Thalidomide and a newer analogue, lenolidamide, are being used therapeutically because they exhibit activity against vascular endothelial growth factor as well as effects on basic fibroblast growth factor activity and immunomodulation.79,80 Lenolidamide is actively being studied in pediatric low-grade gliomas, and thalidomide has been used in a variety of multidrug (metronomic) anticancer regimens. Still another potential biological agent is retinoic acid, which acts as a tumor maturation drug.81 Retinoic acid has effects on both glial and medulloblastoma cells in vitro and is under active study as part of a multiagent approach including more standard chemotherapies in a variety of pediatric brain tumors, especially medulloblastoma. Cell matrix adhesion is an additional target for pediatric brain tumor therapy.82 Integrins are mediators of cell adhesion to the extracellular matrix and play a significant role in the proper migration of cells through both the extracellular and intracellular environment. Integrins also interact with different receptor tyrosine kinases. Cilengitide is an integrin inhibitor that is actively being studied in both pediatric and adult brain tumors. It should be cautioned that the use of these biological agents is extremely complex.6,7 Multiple feedback pathways exist, and it is likely that these agents will have to be used in combination with other biological agents or possibly more standard chemotherapy if they are to play a major role in the treatment of pediatric brain tumors. Specific subtypes of tumors that may have simpler molecular cascades, such as low-grade gliomas, may be more responsive to these biological agents used singly or in simple combinations. How these agents are best used in combination with conventional chemotherapy or radiation therapy is still being elucidated. Given that many of these same pathways are involved in normal brain development, the use of inhibitors has to be closely monitored because of their potential to cause neurodevelopmental toxicities. Among all patients with childhood primary CNS tumors, the greatest experience with the use of chemotherapy has been in those with medulloblastoma. Prospective randomized studies have demonstrated that children with medulloblastoma can be broadly separated into two risk groups. New molecular markers, however, may change risk stratification significantly in the future.1,8,9,83 After treatment with surgery and radiotherapy, children with disseminated disease at the time of diagnosis, and possibly those whose tumors are large and involve the brainstem or are not amenable to total resection, have a poor prognosis, with an overall survival rate after radiation therapy alone of approximately 30 to 40% at 5 years (poor-risk patients). In contradistinction, those patients with localized disease at the time of diagnosis and whose tumors are amenable to aggressive resection have an approximately 50 to 60% survival rate at 5 years (average-risk patients). The addition of chemotherapy during and after radiotherapy has appeared to raise survival rates approximately 20% for both subgroups. This has led to a generation of studies in which children with poor-risk medulloblastoma were treated with chemotherapy more aggressively in an attempt to improve survival, whereas patients with average-risk disease were treated according to protocols combining radiotherapy and chemotherapy, primarily intended to reduce the amount of radiotherapy given in an attempt to decrease the late effects of treatment. There is also a strong rationale for using chemotherapy as primary therapy for some children with medulloblastoma (especially very young children) to allow deferral and, occasionally, avoidance of radiotherapy (▶ Table 50.3). Multiple reports have documented the objective response of recurrent medulloblastoma to a variety of different chemotherapeutic agents, including cisplatin (CPDD), carboplatin, cyclophosphamide, and etoposide, both singly and in combination regimens.84–91 However, despite response rates as high as 80% in some studies, durable survival has been rarely documented after conventional doses of chemotherapy. In addition, there are few, if any, long-term survivors noted in series describing children who failed initial treatment with surgery and craniospinal radiotherapy treated with conventional doses of chemotherapy. High-dose chemotherapy supplemented with autologous bone marrow rescue has been used in children with recurrent medulloblastoma.22,23,92–95 A high rate of response has been documented in such studies; however, preliminary studies also showed a high rate of treatment-related morbidity. For example, the combination of high-dose thiotepa, etoposide, and carboplatin was used in 23 patients with recurrent medulloblastoma.22,23,92 Three patients in this series died of treatment-related toxicity. However, three patients survived without evidence of progression at a median of 36 months from treatment (range, 10 to 63 months). Those patients who did the best after high-dose chemotherapy were those who had minimal residual disease before the use of chemotherapy and no evidence of leptomeningeal dissemination. Other centers, using other high-dose chemotherapeutic regimens, have not reported as many patients with long-term disease control.93–96 It is likely that even if high-dose chemotherapeutic regimens are documented to be efficacious in some children with recurrent medulloblastoma, the autologous bone marrow rescue approach will be replaced by studies using high-dose chemotherapy supported by peripheral stem cell rescue. Biological therapy for patients with recurrent medulloblastoma has primarily focused on the use of sonic hedgehog inhibitors, and responses, although primarily transient, have been noted to date.92 Notch pathway inhibitors have also been used.73,74 Based on studies that demonstrated objective responses to chemotherapy in children with recurrent medulloblastoma, post-surgery trials have been undertaken to evaluate the efficacy of chemotherapy, when added to radiotherapy, for children with newly diagnosed disease (▶ Table 50.4).97–112 Two large prospective randomized trials were performed independently by the Children’s Cancer Group (CCG) and the International Society of Pediatric Oncology (SIOP).1,83 In both studies, patients were randomized to receive radiation therapy alone (3,600 centigray [cGy] of craniospinal irradiation plus local tumor boost to a total dose of 5,400 to 5,600 cGy to the tumor site) or identical radiation therapy plus vincristine (VCR) during the radiation and postradiation therapy cycles of 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) and VCR. For children in the CCG trial, the postradiation chemotherapy regimen also included prednisone. These trials demonstrated for the first time a statistical benefit for the addition of chemotherapy in children with poor-risk posterior fossa medulloblastoma. In the CCG trial, the estimated 5-year progression-free survival (PFS) rate was 59% for children treated with irradiation and chemotherapy and 50% for those who were treated with irradiation alone. However, in patients with a larger tumor bulk at the time of initial surgery and those with the most extensive tumor, there was a demonstrable benefit from the addition of chemotherapy because the event-free survival (EFS) rate was 48% for those receiving chemotherapy, whereas it was 0% for those treated with radiation alone. While these prospective randomized trials were being performed, other single-institution trials were also being completed. McIntosh and colleagues97 reported that 81% of 21 children treated with postradiation therapy cyclophosphamide and VCR were alive and free of disease at a median of 6 years after diagnosis. Packer and colleagues reported that in a three-center trial evaluating 63 children with posterior fossa medulloblastomas, the PFS rate at 5 years was 85.6%.98,99 For eligibility in the latter study, children had to be older than 3 years and must have had subtotal resection, evidence of metastatic disease at the time of diagnosis, and/or brainstem involvement. Patients with metastatic disease at the time of diagnosis had a 5-year PFS rate of 67.15% versus 90.6% for those with localized disease. A study completed by the CCG prospectively treated children who had so-called poor-risk disease with craniospinal and local boost radiotherapy and concomitant VCR chemotherapy during the radiation and eight 6-week cycles of postradiation CCNU and VCR and CPDD, or with 8-drugs-in-1-day therapy for two cycles before irradiation, then craniospinal and local boost radiation, then eight postradiation therapy cycles of 8-drugs-in-1-day therapy.100 The 3-year EFS rate for the group as a whole was 57%. However, those children who received the control arm of CCNU and VCR had a statistically higher 5-year EFS rate than did those who received pre- and postradiation 8-drugs-in-1-day therapy (3-year PFS rates of 62.8% vs. 48.8%). A multicenter randomized trial performed by the SIOP that involved 364 children with biopsy-proven medulloblastoma could not demonstrate a benefit for the use of preradiation chemotherapy with a regimen of methotrexate, procarbazine, and VCR.102 A study performed by the German Cooperative Group, in which an even more aggressive approach to preradiation chemotherapy was used, could not show benefit from treatment with chemotherapy. There was also poorer disease control in the children with localized disease who received preradiation chemotherapy than in those treated with radiation plus postradiation CCNU, VCR, and CPDD chemotherapy.105 In another randomized trial, the Pediatric Oncology Group (POG) compared postradiation nitrogen mustard, VCR, prednisone, and procarbazine as adjuvant therapy with craniospinal irradiation alone.102 The 5-year EFS rate for the group receiving radiotherapy and chemotherapy was 68%, compared with 57% for those receiving irradiation alone. Given the number of patients entered, however, this difference was not statistically significant. The results of another SIOP trial, PNET-3, compared the outcomes of patients who had nondisseminated disease treated with radiotherapy alone (3,600 cGy of craniospinal radiation) with the outcomes of patients who received four cycles of preradiation chemotherapy with oral VP-16, VCR, carboplatin, and cyclophosphamide.108 Overall survival did not differ statistically between the groups, but the EFS rate at 5 years was lower in those who received radiotherapy alone (5-year EFS rate of 74.2% for those receiving radiation plus chemotherapy vs. 59.8% for those who received radiotherapy alone; p = 0.0928). Although prospective randomized studies comparing 3,600 cGy of craniospinal radiotherapy with reduced-dose craniospinal radiotherapy and adjuvant chemotherapy have never been performed, one of the most compelling studies was one by the Children’s Oncology Group (COG) that used 2,340 cGy of craniospinal radiotherapy and either of two chemotherapy regimens: CCNU, CPDD, and VCR or cyclophosphamide, CPDD, and VCR.109 In both treatment arms, VCR was used during radiotherapy. In 379 evaluable patients, the 5-year PFS rate was 86% ± 9%; there was no difference between the two chemotherapeutic regimens.109 A study using higher-dose chemotherapy following radiotherapy, supported by peripheral stem cell rescue, demonstrated a similar rate of PFS.110 A French study also demonstrated that the dose of craniospinal radiotherapy could be reduced to 2,400 cGy if chemotherapy was added.111 These studies, taken in total, suggest that adjuvant chemotherapy is of benefit in children with medulloblastoma. The trials, to date, that have used preradiation chemotherapy have shown no clear advantage for the addition of such chemotherapy in children with more extensive disease at diagnosis and poorer survival for those with average-risk disease compared with immediate postoperative radiotherapy and adjuvant chemotherapy during and after radiotherapy.112 Preradiation chemotherapy may be better than treatment with radiotherapy alone for patients with disseminated disease, but it is unclear whether it is as effective as treatment with radiotherapy plus chemotherapy during or after irradiation. It is paradoxical that children with average-risk disease treated with radiation alone seem to have no better survival rates or, in some studies, poorer survival rates than do children with poor-risk disease treated with radiation plus chemotherapy.108 For children with high-risk medulloblastoma, another potential use of chemotherapy is to intensify treatment, either by administering chemotherapy during radiation (carboplatin) to act as a radiosensitizer or by increasing the dose of postradiation chemotherapy by means of peripheral stem cell rescue. Both approaches have shown possible benefit. Infants are yet another subset of children with medulloblastoma who have been extensively treated with chemotherapy.113,114 Most studies indicate that the prognosis for children in whom medulloblastoma is diagnosed in the first 3 to 4 years of life is poorer than that in older children, independently of whether they are treated with radiation alone or radiation in conjunction with chemotherapy. Because cranial irradiation in young children has been associated with severe adverse delayed toxicities, including intellectual deterioration and endocrinologic (especially growth) sequelae, multiple studies dating back to the 1970s have utilized postsurgical chemotherapy, with delayed or no radiotherapy, for infants with medulloblastoma. In an early study, the use of mechlorethamine, Oncovin (VCR), procarbazine, and prednisone (MOPP) chemotherapy in 13 children younger than 36 months of age with medulloblastoma resulted in a PFS rate of 55%.115 The largest early experience has been that of the POG, which used a four-drug regimen of VCR, cyclophosphamide, CPDD, and etoposide in children younger than 3 years of age with medulloblastoma until they were 36 months of age, or for at least 12 months.5 In this protocol, delayed irradiation was given upon completion of chemotherapy, and the median time to relapse in patients with medulloblastoma was 9 months; 34% of patients remained progression-free for a median of 2 years from diagnosis. No patient had a relapse beyond 26 months from diagnosis, raising the issue of the necessity of irradiation in those with a complete response. The CCG has used the 8-drugs-in-1-day chemotherapy regimen for children younger than 18 months of age with newly diagnosed medulloblastoma.113 In this study, radiation therapy was not routinely employed in those children with a complete response, and the 3-year PFS rate was 22%. The German Cooperative Group has used four cycles of an aggressive regimen of ifosfamide, methotrexate, CPDD, VCR, and intravenous cytosine arabinoside (ara-C) for infants with medulloblastoma.116 The treatment of patients with localized disease at diagnosis resulted in a 2-year disease-free survival rate of 92% without radiotherapy. In contrast, in all patients with disseminated disease, their disease ultimately progressed during or after chemotherapy treatment. In an even more aggressive approach, investigators at Memorial Sloan-Kettering Cancer Center in New York City and participating institutions treated children younger than 3 years who had localized medulloblastoma and children between 3 and 6 years of age who had disseminated medulloblastoma with five cycles of induction chemotherapy (CPDD, high-dose cyclophosphamide, etoposide, and VCR) followed by a consolidation cycle of myeloablative chemotherapy (thiotepa, etoposide, and carboplatin), supported by autologous bone marrow rescue.117 In 13 patients, the 5-year EFS and overall survival rates were 51% and 61%, respectively. Still another approach is the addition of intrathecal chemotherapy to systemic chemotherapy. Infants with nondisseminated medulloblastoma treated with high-dose intravenous and intraventricular methotrexate and cytosine arabinoside, coupled with other systemic chemotherapeutic agents, demonstrated a nearly 60% 5-year PFS rate without receiving radiotherapy.118–120 This study and a subsequent follow-up study achieved a disease-free survival rate of greater than 80% for those with nodular or desmoplastic medulloblastoma, demonstrating both the better prognosis for the subgroup of patients with that variant of medulloblastoma and that some infants can be treated with chemotherapy alone after surgery. Methotrexate has been associated with a high rate of leukoencephalopathy, raising issues over the long-term benefits of such approaches. The ability of chemotherapy alone to control disseminated disease in infants has never been shown. Other approaches are coupling biological agents, including retinoic acid and SAHA (suberoyl anilide hydroxamic acid), with multiagent chemotherapy to improve disease control. The chemotherapy used for children with primitive neuroectodermal tumors (PNETs) arising outside the posterior fossa, including pineoblastomas, has mirrored that used for children with medulloblastomas. Data suggest that children with nonposterior fossa PNETs have a poorer prognosis than those with medulloblastomas, possibly because of their younger age at the time of diagnosis or because such tumors (especially pineoblastomas) are frequently disseminated early in the course of illness.121–123 Also, non–posterior fossa tumors are genomically different from medulloblastomas. The role of adjuvant chemotherapy for children with non–posterior fossa PNETs has not been well demonstrated.121,122 This is partially due to the relatively small numbers of patients available for study. In the CCG trial comparing pre- and postradiation chemotherapy with the 8-drugs-in-1-day regimen versus treatment with radiation therapy plus CCNU and VCR, children with both pineal PNETs and non–posterior fossa, nonpineal PNETS were treated. For the 44 patients with supratentorial PNETS, the PFS rate was 45.8%. In addition, 25 children with pineal tumors were treated. Eight were younger than 18 months and were nonrandomly treated with the 8-drugs-in-1-day chemotherapy regimen. The remaining 17 patients were randomized between the two treatments. All infants in this study developed progressive disease at a median of 4 months from the start of treatment. In the 17 older patients, the overall 3-year PFS rate was 61.13%. The numbers in this study are too small to evaluate the relative efficacy of either of the two adjuvant chemotherapy regimens. In the SIOP study, 25 children with supratentorial PNETs were treated with chemotherapy alone, and 24 had a relapse at a median of 5.5 months. In a study in older children, radiotherapy plus CCNU, VCR, and CDDP chemotherapy resulted in an approximately 50% rate of disease control.124 High-dose chemotherapy with peripheral stem cell rescue using cyclophosphamide, CPDD, and VCR following radiotherapy has also been tried in this population, with at least evidence that such an approach is feasible.19 Given the poor outcome of children with non–posterior fossa PNETs, most investigators are using some form of chemotherapy in addition to radiation therapy. The issue of the efficacy of chemotherapy for malignant gliomas in childhood remains somewhat unsettled.3 To date, chemotherapeutic trials for pediatric malignant gliomas have largely been initiated based on the encouraging results observed for a particular drug or regimen first investigated in adult malignant gliomas. However, it is now becoming clear that adult GBMs are molecularly heterogeneous and quite distinct from pediatric lesions.125,126 Thus, it is very likely that future pediatric trials will need to be designed and tailored more specifically for the molecular targets uniquely represented in the pediatric form of the disease. The molecular genetic alterations of both childhood and adult malignant gliomas are still being elucidated, but emerging evidence now demonstrates that malignant astrocytic tumors arising in childhood are more likely to have TP53 mutations and thus a genetic composition different from that of the neoplasms of adulthood, which are believed to go through a cascade of genetic changes as they progress from low-grade to high-grade malignancies.127 This may in part explain the differences, if they exist, between the sensitivities to chemotherapy of adult and pediatric malignant gliomas. For example, adult patients with GBM demonstrated a clear survival benefit with the addition of temozolomide to standard-dose irradiation; however, a similar benefit was not demonstrated in an identical pediatric malignant glioma trial.128 Molecularly targeted therapy against alterations more commonly observed in high-grade pediatric gliomas, such as PDGFRA amplification, are under way at some centers. Other strategies, such as the combination of anti-angiogenesis inhibitors or other novel biological agents with radiation, are being investigated. Clinical trials, as noted in ▶ Fig. 50.1, are ongoing to better define the role of chemotherapy in high-grade glioma management. At the time of recurrence, malignant gliomas may transiently respond to chemotherapeutic agents. Interpretations of chemotherapy trials are difficult, especially in adult series, because many trials combine patients with objective neuroradiographic response together with those who have disease stabilization, considering both groups as “responders.”129–131 The nitrosoureas singly or in combination with other drugs, such as procarbazine and VCR, have been the most extensively studied drugs in adult Phase II trials; overall objective response rates usually have ranged from 10 to 20% (although some report transient benefit in as many as 75% of patients). Median time to progression in most series, which may be a better marker of efficacy, is usually less than 6 months, with most series documenting disease progression by 20 to 25 weeks after the initiation of treatment. Studies in purely pediatric populations with recurrent malignant gliomas have been undertaken less commonly. Drugs that have been evaluated include VCR, procarbazine, intravenous etoposide, oral etoposide, BCNU, CCNU, CPDD, cyclophosphamide, carboplatin, irinotecan, and thiotepa as single agents or in combination with another agent, such as ifosfamide or high-dose cytosine arabinoside.132–150 These studies have shown objective response rates ranging from 0 to 50%, with most studies reporting tumor shrinkage in 10% of patients or fewer. As in adult trials, even in those series reporting a higher rate of response, the median time to progression has been less than 1 year and usually 6 months or less. Temozolomide is an oral alkylating agent that has been well tolerated and has shown promising but mixed results in adults and children with recurrent high-grade gliomas.151–153 In adult trials, objective responses plus stable disease were reported in as many as 67% of patients with recurrent grade II tumors. For patients with GBM, similar overall “responses” were noted, but the duration of response was shorter. Experience in pediatric patients has been less extensive, with a greater variability noted in pediatric series, with objective responses ranging from 0% in a CCG series to as high as 50% in a smaller limited-institution trial.151 One approach to increase the efficacy of temozolomide and other drugs that alkylate DNA in the O6 position, such as the nitrosoureas and procarbazine, is to couple such treatments with drugs that bind to the DNA repair protein O6-alkylguanine DNA-alkyltransferase. One such drug is O6-benzylguanine. Despite encouraging preliminary data, the most recent pediatric clinical trial for refractory high-grade gliomas using the combination of temozolomide and O6-benzylguanine failed to demonstrate efficacy.154 There has been interest in the use of higher doses of chemotherapy for children with malignant gliomas supplemented by either autologous bone marrow rescue or peripheral stem cell rescue.22,155–157 Thiotepa and etoposide have been used at high doses for children with malignant gliomas. Objective responses to therapy were noted in 6 of the first 10 patients treated. Later studies combining high-dose BCNU with thiotepa and etoposide or high-dose carboplatin with thiotepa and etoposide disclosed a similar overall response rate in a larger group of patients. These studies also encouragingly disclosed a small subgroup of children with prolonged survival, including children with anaplastic gliomas and those with GBMs. These results were tempered by a toxic mortality rate of nearly 20% in the preliminary studies. Other groups using different chemotherapeutic agents, such as busulfan and thiotepa or melphalan-based regimens, have not been able to demonstrate such high response rates or prolonged responses to chemotherapy.22,155–157 The reasons for these disparities in studies are unclear; however, it does seem that patients with minimal residual disease before treatment with high-dose chemotherapy (those with tumors that could be debulked before chemotherapy) were the most likely to derive a prolonged benefit from treatment. Despite multiple single-institution trials and prospective randomized studies, the value of adjuvant chemotherapy for adults with malignant gliomas is far from dramatic. Chemotherapy for adults, when given as an adjuvant with or after radiation therapy, produces a modest prolongation in median survival but has not clearly been shown to improve the likelihood of long-term survival.158–163 A meta-analysis of major adjuvant chemotherapy trials concluded that there was a 10% increase in survival at 1 year and an 8.6% survival advantage at 2 years for adults who had GBM treated with chemotherapy and radiotherapy compared with those treated with irradiation alone.164,165 The study that has suggested most strongly that adjuvant chemotherapy is of benefit for children with high-grade gliomas was completed by the CCG in 1982.3 In this trial, the addition of CCNU and VCR chemotherapy, during and after radiotherapy, increased survival compared with treatment with radiation alone. Of the children in this randomized trial who received radiotherapy and adjuvant chemotherapy, 46% were alive and free of disease 5 years following treatment versus 18% of those treated with postsurgery radiotherapy alone. The benefit of chemotherapy was statistically significant for children with GBM. In a follow-up study, the CCG compared pre- and postradiation chemotherapy with the 8-drugs-in-1-day regimen versus therapy with irradiation and CCNU and VCR.166 No survival advantage was shown for the children treated with pre- and postradiation 8-drugs-in-1-therapy in comparison with those treated with adjuvant CCNU and VCR. Overall, the survival rates for children with anaplastic gliomas and those with GBM were somewhat lower in the most recent CCG trial, but approximately 30% of children with anaplastic gliomas and 20% of children with GBM were alive and free of disease 5 years following treatment. A statistical comparison of these two trials showed no difference in overall survival between them.167 A more recent review of this information suggested that some of the children considered to have high-grade gliomas in the first CCG trial actually had low-grade gliomas. However, even when the pathologic materials were reviewed again, there was a statistical benefit for the addition of CCNU and VCR for children with GBM. Given the equivocal results of adjuvant trials and the preliminary results of high-dose chemotherapy for recurrent high-grade glioma, studies are presently ongoing using high-dose chemotherapy either before or following radiation therapy for children with anaplastic gliomas (primarily subtotally resected tumors) and children with GBM. A CCG trial that used couplets of agents, including cyclophosphamide and VCR, ifosfamide and VCR, and carboplatin and VCR, before irradiation demonstrated an overall poor objective response rate of less than 20%.168 Alternatively, trials of chemotherapy during radiation therapy with agents like temozolomide in patients with newly diagnosed disease are ongoing. However, temozolomide alone given concurrently with radiation on a daily low-dose schedule followed by a standard dose and scheduled maintenance temozolomide failed to improve survival in the most recent COG trial for newly diagnosed pediatric malignant gliomas.128 Newer biological agents, such as inhibitors of tyrosine kinase receptors (anti-EGFR and anti-PDGFR), epigenetic modifying agents such as histone deacetylase (HDAC) inhibitors, anti-angiogenesis agents (e.g., bevacizumab, cilengitide), and cell signal–disrupting agents, have also been coupled with irradiation in attempts to improve disease control. The majority of children with brainstem gliomas have diffuse infiltrating lesions that primarily involve the pons and result in death within 18 months of diagnosis. Because of this poor survival rate, chemotherapy has been used in children with both newly diagnosed and recurrent brainstem gliomas. Such studies are still ongoing, but there has been increased interest in using chemotherapy during radiotherapy for patients with newly diagnosed disease. At the time of recurrence, a variety of single agents investigated in patients with high-grade gliomas (e.g., intravenous carboplatin, cisplatin, and etoposide; oral thiotepa and CCNU) have resulted in responses in only 0 to 20% of patients.169–173 Temozolomide has also been used, with disappointing results. Furthermore, in the majority of cases, even if partial tumor shrinkage occurred, the response was relatively short. Oral etoposide resulted in a response in 4 of 12 children with recurrent brainstem gliomas.169 In addition, a variety of drug combinations have been tried without clear benefit. Adjuvant postradiation chemotherapy has been poorly studied in children with brainstem gliomas. In one of the few randomized prospective studies performed to date, the addition of postradiation CCNU and VCR did not improve the length or frequency of disease-free survival in comparison with radiotherapy alone.174 The median PFS in this study of 79 children with brainstem gliomas was 7 months for those receiving radiation therapy alone and 6 months for those treated with radiation and chemotherapy. Chemotherapy has been used before irradiation in an attempt both to identify active agents and to improve survival. One study of cisplatin and cyclophosphamide in 32 children demonstrated that such therapy could be delivered before radiotherapy but resulted in a poor overall response rate (3 of 32, or 9%) with no improvement in survival.175 Carboplatin given before radiotherapy resulted in responses in 2 of 27 patients; the median overall survival in this series was 9 months.171 In a CCG trial, treatment with cisplatin, etoposide, cyclophosphamide, and VCR or with carboplatin, etoposide, and VCR before irradiation resulted in few objective responses and no apparent improvement in survival.176 Another trial coupled carboplatin with a bradykinin agonist during radiotherapy to improve drug delivery but did not show apparent improved survival. High-dose, multiagent chemotherapy, such as that used in protocols designed for patients with high-grade cortical gliomas, has been attempted in this population before irradiation. However, because of inordinate toxicity and a lack of clear efficacy, these studies were aborted.173–175 Other studies are evaluating the efficacy of chemotherapy, biological agents, and novel radiosensitizers during and after radiation in attempts to improve survival. To date, no agent has shown clear benefit. Interferon has been used in children with both recurrent and newly diagnosed brainstem gliomas.176–178 Nagai and Arai177 demonstrated objective responses in 8 of 20 patients with recurrent high-grade gliomas after intrathecal or intratumoral infusion of interferon-β. Interferon-β resulted in objective tumor responses in 2 of 9 children with recurrent brainstem gliomas and prolonged disease stabilization in 2 patients in another series.176 However, a follow-up study of interferon-β, given both during and following irradiation for patients with newly diagnosed brainstem gliomas, did not demonstrate increased efficacy.178 In this latter study, 32 children were treated, and 30 of 32 developed progressive disease at a median of 5 months from diagnosis. In contrast, a study by Wakabayashi et al179 treated 16 pediatric patients who had a diagnosis of brainstem glioma with interferon-β, ACNU ([1-(4-amino-2-methyl-5-pyrimidinyl) methyl-3-(2-chloroethyl)-3-nitrosourea hydrochloride]), and radiation therapy. Of 8 patients in this series with diffuse intrinsic tumors, 7 were reported to have a complete or partial response to treatment, with a median overall survival of 15.7 months. Surprisingly, in the study of Wakabayashi et al, the best survival rate was seen in patients with diffuse intrinsic tumors. The role of interferon-β for patients with brainstem gliomas thus remains unclear, but studies continue to be performed with this agent and other forms of immunotherapy. Low-grade gliomas constitute the majority of childhood brain tumors. Until the 1980s, chemotherapy was used sparingly in children with recurrent and newly diagnosed low-grade gliomas.180,181 Most studies were performed primarily in children with diencephalic tumors. Similar outcomes, in smaller numbers of patients, have been reported in children with tumors in other CNS sites, including the brainstem. For exophytic pilocytic astrocytomas, a variety of drugs demonstrated efficacy against recurrent tumors, although in most cases, these regimens were used in only a small group of patients.181–187 Drugs or drug combinations that have been used and found to be somewhat effective in children with recurrent low-grade gliomas include actinomycin and vincristine, carboplatin alone, carboplatin and vincristine, vinblastine, vinblastine and carboplatin, etoposide, etoposide and cisplatin, temozolomide, and multidrug regimens, such as TPCV (CCNU, procarbazine, vincristine, 6-thioguanine, and dibromodulcitol). Most studies using single-agent temozolomide, vinblastine, or carboplatin reported stable disease, although the regimen of carboplatin and vincristine resulted in objective tumor shrinkage in 9 of 19 patients.186 Biological agents have recently been incorporated into the therapy of recurrent low-grade gliomas. Bevacizumab plus irinotecan demonstrated a 30 to 60% response rate, at times associated with visual or neurologic improvement, in children with multiply recurrent low-grade gliomas.61 Drugs targeting the RAS/MAPK pathway are being actively explored, stimulated by the discovery of BRAF mutations in the majority of children with pilocytic astrocytomas.62 Inhibition of BRAF and MEK, as well as inhibitors of mTOR, are under active study. The role of chemotherapy for children with newly diagnosed low-grade gliomas is established, although the optimal regimen remains to be determined. In attempts to decrease the potential long-term sequelae of radiation therapy, chemotherapy has been used to at least delay, if not obviate, the need for radiation therapy. The largest early experience with chemotherapy was with the combination of actinomycin D and VCR.188 In a series of 30 patients younger than 5 years of age with newly diagnosed, progressive visual pathway gliomas, 80% of the children were shown to have at least disease stabilization while receiving chemotherapy (an overall objective response was seen in 20% of patients). This stabilization lasted for a mean of 3 years and resulted in the requirement for radiation at a median age of 4.5 instead of 1.5 years. Subsequently, the combination of carboplatin and VCR has been widely used for children with newly diagnosed or progressive low-grade gliomas at any site in the nervous system.187 The overall response rate after treatment with carboplatin and VCR was 60% (▶ Fig. 50.1). More than 90% of patients, independently of the location of their tumor in the nervous system, experienced at least disease stabilization with this regimen. In a series of 78 patients, the PFS rate with the two-drug regimen was 68% at 3 years, and therapy was shown to benefit both patients with diencephalic tumors and those with progressive low-grade brainstem tumors. Fig. 50.1 Sagittal magnetic resonance imaging after gadolinium (a) at diagnosis, (b) following surgery, (c) at 3 months, and (d) at 12 months after treatment with carboplatin and vincristine in an 11-month-old child without neurofibromatosis with a diencephalic pilocytic astrocytoma.
50.1 General Considerations
50.2 Types of Drug Trials
Drug
Route
Tumor type
More common toxicities
Alkylating agents
Cyclophosphamide
IV
Medulloblastoma Germ cell tumors
Myelosuppression Hemorrhagic cystitis
Ifosfamide
IV
Medulloblastoma
Hemorrhagic cystitis Reproductive
Melphalan
IV (ABMR)
? Medulloblastoma ? High-grade glioma
Myelosuppression Gastrointestinal
Thiotepa
IV (ABMR)
? Medulloblastoma ? High-grade glioma
Myelosuppression
CCNU (BCNU)
PO (IV)
Medulloblastoma High-grade glioma Oligodendroglioma
Myelosuppression Gastrointestinal Pulmonary
Cisplatin
IV
Medulloblastoma ? Ependymoma Germ cell tumors
Ototoxicity Nephrotoxicity Myelosuppression Peripheral neuropathy Nausea and vomiting
Carboplatin
IV
Low-grade glioma Medulloblastoma ? Ependymoma Germ cell tumors
Peripheral neuropathy Nausea and vomiting Myelosuppression
Procarbazine
PO
? High-grade glioma ? Low-grade glioma ? Oligodendroglioma
Myelosuppression Nausea and vomiting
Temozolomide
PO
? High-grade glioma ? Low-grade glioma ? Other
Myelosuppression Hepatotoxicity
Antimetabolites
Methotrexate
PO, IV, ? IT
? Medulloblastoma ? High-grade glioma
Myelosuppression Gastrointestinal Hepatotoxicity
Cytosine arabinoside
IV, ? IT (? ABMR)
? Medulloblastoma
Gastrointestinal Hepatic
Plant alkaloids
Vincristine
IV
Medulloblastoma Low-grade glioma ? High-grade glioma
Peripheral and autonomic neuropathy
Etoposide (VP-16)
IV or PO
? Medulloblastoma ? High-grade glioma ? Low-grade glioma Germ cell tumors
Myelosuppression
Abbreviations: ABMR, autologous bone marrow rescue; BCNU, bischlorethyl-nitrosourea; CCNU, 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea; IT, intrathecal; IV, intravenous; PO, orally.
Type
Goal
Patient population
Phase I
Determine maximum tolerated dose; ? dose searching
Recurrent tumors without known effective treatments; 15 to 30 patients
Phase II
Determine spectrum of agent; estimate disease-specific response rate
Recurrent tumors; newly diagnosed refractory tumors; 15 to 30 patients per tumor type
Phase III
Determine if more effective than currently available therapy
Usually newly diagnosed; often randomized
50.3 Determinants of Drug Efficacy and Drug Resistance
50.4 Biological Therapy
50.5 Treatment of Specific Tumor Types with Chemotherapy
50.5.1 Medulloblastoma
Tumor type
Study type
Chemotherapy
Radiotherapy
Average-risk medulloblastomas
Phase III
During and post-RT: VCR, CPDD, CCNU, Cyclo, VP-16
CSRT: 2,400 cGy versus 1,800 cGy; 5,500 cGy posterior fossa versus tumor site RT
Poor-risk medulloblastomas
Phase III
Carbo during RT versus no Carbo with and without retinoic acid
CSRT: 3,600 cGy; 5,940 cGy local RT
Infantile malignant tumors
Phase I/II
High-dose chemotherapy with Cyclo, VCR, CPDD, VP-16, and thiotepa/carboplatin with PSCR with and without MTX
? Post-Rx RT
Low-grade gliomas
Phase III
Carbo/VCR versus CCNU, PCB, 6-TG, VCR
No RT
Low-grade gliomas
Phase II
Carbo/VCR/temozolomide
No RT
Ependymomas
Phase II
Pre-RT, second-look surgery: Carbo, VCR, VP-16, CPDD, Cyclo
Local RT
Ependymomas
Phase III
Carbo, VP-16, Cyclo versus no chemo for anaplastic post-RT
Local RT
High-grade gliomas
Phase II
Pre-RT chemotherapy/ biological therapy
Local RT
Nongerminomatous germ cell tumors
Phase II
Pre-RT ifosfamide, Carbo, VP-16, ifosfamide, thiotepa
?
Abbreviations: Carbo, carboplatin; CPDD, cisplatin; CSRT, craniospinal RT; Cyclo, cyclophosphamide; MTX, methotrexate; PCB, procarbazine; PSCR, peripheral stem cell rescue; RT, radiotherapy; 6-TG, 6-thioguanine; VCR, vincristine; VP-16, etoposide.
Recurrent Disease
Newly Diagnosed Disease
Study (No. of patients)
Type of trial
Dose of CSRT (cGy)
Drugs used
Outcome
McIntosh et al97 (21)
Single arm; all risks
WB 3,925; spinal 3,300
VCR; Cyclo
PFS 5 years 81%
Evans et al1 (179)
Randomized; all risks
WB 3,600; spinal 3,600
CCNU/VCR; prednisone versus RT alone
PFS 5 years 60%, w/CHT (high-risk 46% CHT versus 0% CHT)
Tait et al83 (251)
Randomized; all risks
WB 3,500–4,500; spinal 3,000–3,500
CCNU; VCR versus RT alone
PFS 5 years 53%
Bailey et al102 (364)
Randomized; all risks
WB 3,500; spinal 3,500 versus WB 2,500; spinal 2,500
Pre-RT VCR; PCB; MTX versus VCR; CCNU post-RT
EFS 5 years pre-RT plus no post-RT 2,500 cGy 41% ± 8%
Packer et al98,99 (63)
Prospective; nonrandomized; high-risk
WB 3,600/2,400; spinal 3,600/2,400
Post-RT CCNU; VCR; CPDD
PFS 5 years 85% ± 6%
Packer101 (65)
Prospective; nonrandomized; average-risk
WB 2,340; spinal 2,340
Post-RT CCNU, VCR; CPDD
PFS 5 years 79% ± 7%
Zelzer et al100 (203)
Randomized; high-risk
WB 3,600; spinal 3,600
Pre RT 8-in-1 versus post-RT CCNU; VCR
PFS 5 years 63% ± 5% Post-RT versus 45% ± 5% pre-RT
Kortmann et al105 (156)
Randomized; all risks
WB 3,520; spinal 3,520
Pre-RT Ifos; VP-16; MTX, CDDP; ara-C versus VCR; CCNU; CPDD post-RT
EFS 3 years 65% pre-RT versus 78% post-RT
Taylor et al107 (217)
Randomized; average-risk
WB 3,500; spinal 3,500
Pre-RT VCR; VP-16; Carbo; Cyclo
EFS 5 years 71.6% (74:2% for pre-RT versus 54.8% for RT)
Packer at al109 (420)
Randomized; average-risk
CSRT 2,340
Post-RT Cyclo, CPDD, VCR versus CCNU, CPDD, VCR
EFS 5 years 81% ± 2% OS 86% ± 9%
Carrie et al111 (136)
Average-risk
CSRT 2,500
Pre-RT 8-in-1; post-RT Carbo/VP-16
PFS 5 years 73.8% ± 7.6%
Gajjar et al110 (134)
All risks
Risk-adapted: 2,340 for average-risk; 3,600–3,960 for high-risk
Post-RT Cyclo; CPDD; VCR plus stem cell
PFS 5 years 85% average-risk; 70% high-risk
Abbreviations: 8-in-1,8-drugs-in-1-day therapy; ara-C, cytosine arabinoside; Carbo, carboplatin; CCNU, 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea; CDDP, CPDD, cisplatin; CHT; CSRT, craniospinal radiotherapy; Cyclo, cyclophosphamide; EFS, event-free survival; Ifos, ifosfamide; MOPP, mechlorethamine, vincristine, prednisone, procarbazine; MTX, methotrexate; OS, overall survival; PFS, progression-free survival; RT, radiotherapy; PCB, procarbazine; post-RT, after RT; VCR, vincristine; VP-16, etoposide; WB, whole brain.
Treatment of Infants
50.5.2 Other Primitive Neuroectodermal Tumors
50.5.3 High-Grade Gliomas
Recurrent Disease
Newly Diagnosed Disease
50.5.4 Brainstem Gliomas
Recurrent Disease
Newly Diagnosed Disease
50.5.5 Low-Grade Gliomas
Newly Diagnosed Disease
Chemotherapy and Biologic Therapy for Pediatric Brain Tumors
Only gold members can continue reading. Log In or Register to continue







Full access? Get Clinical Tree


