Childhood and Juvenile Absence Epilepsies
Edouard Hirsch
Pierre Thomas
Chrysostomos P. Panayiotopoulos
Introduction
Typical Absences: The Symptoms
Typical absences (TAs) are epileptic seizures manifested by impairment of consciousness and 2.5- to 4-Hz generalized spike-and-slow-wave discharges47,85,187 (see Chapter 49). Impairment of consciousness may be mild (requiring special testing)1,7,27,112,122,161,162,199,205,206,208 or severe and may be associated with other clinical manifestations, such as automatisms,184 regional or widespread myoclonia (rhythmic or random), and autonomic disturbances.46 Furthermore, the electroencephalographic (EEG) discharge may be brief or long, continuous or fragmented, with multiple or single spikes that are consistently associated or not associated with the slow wave. The intradischarge frequency may be relatively constant or vary.161
Thus, the term typical absences does not refer to a stereotyped symptom, but to a cluster of clinico-EEG manifestations that may be syndrome related. It should be appreciated that like any other physical symptoms in medicine, a detailed study of the manifestations of TAs is a prerequisite for a meaningful syndrome-related diagnosis. The clinico-EEG manifestations of absences have been best described in the eminent video-EEG studies by Penry et al.185 and Stefan et al.,211 but it is only recently that an attempt has been made for their syndrome-related characterization with video-EEG analysis.108,109,158,159,160,161,170,171,172 It has been shown that some of the manifestations of TA may be more specifically related to an epileptic syndrome than others, but no single symptom is sufficient to define an epileptic syndrome.173
Epileptic Syndromes with Typical Absences
An epileptic syndrome, by definition, requires the nonfortuitous clustering of many symptoms and signs.48
Four epileptic syndromes with TAs have been recognized by the International League Against Epilepsy (ILAE)48: Childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME), and myoclonic absence epilepsy (MAE). The first three (CAE, JAE, JME) are considered as part of idiopathic generalized epilepsies (IGEs), whereas the fourth (MAE) is categorized among the symptomatic or cryptogenic generalized epilepsies.
There may be more epileptic syndromes with TAs, such as eyelid myoclonia with absences (EMA), perioral myoclonia with absences, and others awaiting further studies and confirmation.166,168,171,177,178 Furthermore, idiopathic generalized tonic–clonic seizures on awakening are often associated with mild absences.175 Many of these syndromes are different in presentation, severity, and prognosis. Children with CAE in their majority will remit, those with MAE are affected by or may develop mental and behavioral problems, and those with JME in their midteens may develop lifelong myoclonic jerks and generalized tonic–clonic seizures (GTCSs). Other patients may have subtle clinical manifestations during the typical 3-Hz spike-and-wave discharges of which they are not aware (phantom absences); often they seek medical consultation only after a generalized tonic–clonic seizure develops, probably a long time after the onset of absences.
Historical Perspectives
This topic has been discussed in detail elsewhere.66,67,116,125,127,134,135,139,194 According to Temkin,216 the first description of absences was made in 1705 by Poupart. Tissot in 1770217 described a girl with absences “avec un très léger mouvement dans les yeux” associated with frequent GTCSs. The term epileptic absences was first used by Calmeil35 in his doctoral thesis of 1824. The term of petit mal was introduced by Esquirol in 1815.73 Gowers in 188197 gave the most accurate description of absence seizures “without conspicuous convulsions,” and Hughlings Jackson in 1879115 discussed the differences between absences and complex partial seizures. Absences also have been described on clinical grounds, without EEG, by Friedmann.83 Although he believed that these absences were not epileptic, he gave an excellent description of the attacks and a long-term favorable prognosis. Sauer204 originated the term pyknolepsy (from the Greek pyknos indicating closely packed, dense, aggregated). Adie.2 based on his own observations but mainly, as he admitted, on those of Friedmann,83 Heilbronner,106 and Stier,212 defined pyknolepsy in the most admirable way as follows:
“A disease with an explosive onset between the ages of 4 and 12 years, of frequent short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months or years, and which are uninfluenced by anti-epileptic remedies, do not impede normal and psychical development, and ultimately cease spontaneously never to return. At most the eyeballs may roll upwards, the lids may flicker and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations and lassitude or confusion after the attacks, are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present they should be considered as foreign to the more favourable disease.”
The electroclinical characteristics of absences were described by Gibbs.94,95 The petit mal triad of Lennox,126,127 which was misused and misunderstood, was finally clarified by the ILAE47 with the differentiation of typical and atypical absences.
The present ILAE distinction48 between CAE and JAE is mainly based on the pioneer work of Doose et al.61 and Janz.116 Doose et al.,61 in a study of 149 children with absences, found three different groups with emphasis at the age of onset: (a) an absence epilepsy of early onset from birth to 4 years of age, (b) CAE (pyknolepsy) with onset at 4 to 8 years of age, and (c) JAE with onset before puberty and with absences occurring in clusters (cycloleptic) or sporadically (spanioleptic). Janz116 emphasized the significance of the frequency of the absences in a comparative study of 505 pyknoleptic and 197 nonpyknoleptic cases and confirmed Doose’s conclusions regarding differences of age at onset and sex.116,118,119
Definitions
Childhood Absence Epilepsy
According to the international classification of epilepsies,48 CAE represents an idiopathic generalized epilepsy defined as follows: Pyknolepsy occurs in children of school age (peak manifestation age 6 to 7 years), with a strong genetic predisposition in otherwise normal children. It appears more frequently in girls than in boys. It is characterized by very frequent (several to many per day) absences. The EEG reveals bilateral, synchronous symmetric spike-waves, usually 3 Hz, on a normal background activity (Fig. 1). During adolescence, generalized tonic–clonic seizures often develop. Otherwise, absences may remit or, more rarely, persist as the only seizure type.
This brief definition of the ILAE Commission48 mainly based on retrospective studies was a source of confusion. Thus, many authors make the arbitrary interpretation that CAE is any type of epilepsy with onset of absences in childhood. Therefore, epidemiology, genetics, age at onset, clinical manifestations, other types of seizures, long-term prognosis, and treatment do not accurately reflect the syndrome of CAE. A more precise definition of childhood absence epilepsy has been recently proposed by the ILAE Task Force on Classification defining inclusion and exclusion criteria.140 It takes into account several important diagnostic points, such as the degree of impairment of consciousness, the morphology of spike-wave discharges, and the place of generalized tonic–clonic seizures. Clear exclusion criteria were also proposed140: Eyelid myoclonia (which is predominantly myoclonic and has minimal consciousness impairment) and TAs consistently provoked by specific stimuli. The same applies for multiple spikes (more than three spikes per wave) that also indicate a bad prognosis and coexistent myoclonic jerks or GTCSs.160
The following definition may better represent CAE (Table 1):
Childhood absence epilepsy is an age-related idiopathic generalized epilepsy, which occurs in otherwise normal children, more frequently girls, with a strong genetic predisposition. Age of onset is between 4 and 10 years of age, with a peak at 5 to 7 years. Absences are frequent, tens to hundreds per day. Their duration varies from 4 to 20 seconds, though most of them last around 10 seconds. Clinically, there is abrupt and severe impairment (loss) of consciousness, with cessation of voluntary activity, which is not restored during the ictus. The eyes spontaneously open; overbreathing, speech, and other voluntary activity stop within the first 3 seconds from the onset of the discharge. Automatisms are frequent but have no significance in the diagnosis. The eyes stare or move slowly, and random eyelid blinking (usually not sustained) may occur.
Persistent eyelid myoclonia, rhythmic massive limb jerking, and single or arrhythmic myoclonic jerks of the head, trunk, or limbs are probably not compatible with childhood absence epilepsy. However, milder myoclonic elements, particularly at the onset of the seizure discharge, may be a feature of childhood absence epilepsy. Generalized tonic–clonic seizures and other types of seizures like myoclonic jerks should not be featured in childhood absence epilepsy. Visual (photic) and other sensory precipitation are most likely against a diagnosis of childhood absence epilepsy. Mild or no impairment of consciousness is not compatible with childhood absence epilepsy.
The EEG has a normal background, with sometimes rhythmic posterior delta activity. Ictal discharges consist of generalized high-amplitude spike- and double (maximum occasional three spikes are allowed)-spike-and-slow-wave complexes. They are rhythmic at around 3 to 4 Hz (>2.5 Hz) with a gradual and regular (0.5 to 1 Hz) slowdown from the initial to the terminal phase of the discharge. The first 1 to 2 seconds of the onset of the discharge is usually fast and unreliable for these measurements. There are no marked variations in the relation of spike to the slow wave, no fluctuations in the intradischarge frequency, and certainly no fragmentations of the ictal discharges.
Remission usually occurs before the age of 12 years but infrequent GTCSs may develop in adolescence.
Juvenile Absence Epilepsy
According to the Revised International Classification of Epilepsies and Epileptic Syndromes,48 JAE is one of the age-related idiopathic generalized epilepsies. The following description is given:
The absences of JAE are the same as in pyknolepsy, but absences with retropulsive movements are less common. Manifestation occurs around puberty. Seizure frequency is lower than in pyknolepsy, with absences occurring less frequently than every day, mostly sporadically. Association with GTCS is frequent, and GTCSs precede the absence manifestations more often than in childhood absence epilepsy, often occurring on awakening. Not infrequently, the patients also have myoclonic seizures. Response to therapy is excellent.
A similar but slightly more restrictive definition has been proposed by Panayiotopoulos.166
Epidemiology
Childhood Absence Epilepsy
The annual incidence of CAE is low and may vary from 1.9 to 8 per 100,000 children below the age of 16 years, and the prevalence is probably in the range of 2% to 10% of children with epileptic disorders.23,30,133,200,201 A twofold preponderance in girls than boys may be a realistic estimate, although some studies have reported that boys and girls are equally affected.
Juvenile Absence Epilepsy
There are no population-based epidemiologic data on this syndrome. According to Janz,118 JAE represented 10% of the age-related epilepsies with petit mal seizures.
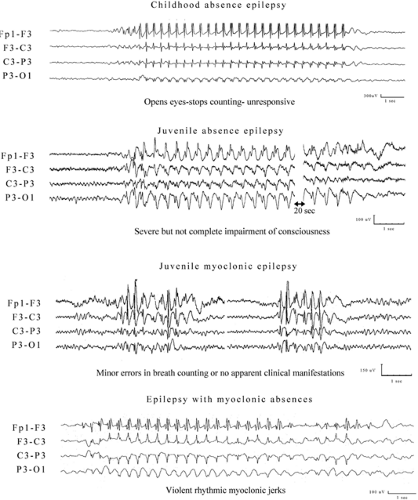 FIGURE 1. Video-electroencephalographic (EEG) recordings. Upper: This trace is from a 7-year-old girl with frequent daily typical absences (TAs). She stops overbreathing and counting within the first 3 seconds after onset of the EEG 3-Hz spike-and-slow-wave discharge, she is unresponsive, and she demonstrates features described in the text for CAE. Automatisms are seen in some of her seizures, but there is no eyelid or perioral or limb myoclonia. She is not photosensitive. Note the regularity and rhythmicity of the ictal paroxysm. Upper middle: This trace is from a 33-year-old woman who has experienced frequent daily absences that were highly resistant to treatment since 8 years of age. Ten long spontaneous absences were recorded during the 3-hour video-EEG session, but they were mainly provoked by overbreathing. She experiences severe impairment of consciousness, but she may recall events occurring toward the end of the ictus, usually keeps her eyes closed, and may restore counting during the discharge. The long ictal EEG is as rhythmic and regular as that of CAE. Lower middle: This image is from a 38-year-old woman with juvenile myoclonic epilepsy (JME). Note the brief fragmented discharges with “W’s. Impairment of consciousness may be detected with breath counting (annotated with numbers) where there is a significant delay in pronouncing the next number after a discharge. Lower: This trace is from an 11-month-old boy with mild developmental delay and a 1-month history of absences. In video-EEG these were manifested with 3-Hz rhythmic multiple spike-and-slow-waves with rhythmic myoclonic jerking of the head, body, and shoulders. |
Table 1 Inclusion and Exclusion Criteria for Childhood Absence Epilepsy | |||||
|---|---|---|---|---|---|
|
Etiology and Basic Mechanisms
According to Tissot,217 “To produce epilepsy, two things are necessary: (i) a tendency for the brain to fall into spasm more readily than during health; (ii) a source of irritation that can precipitate this tendency.” These two factors (genetic factor and acquired factor) exist with a very unequal significance in absences epilepsies.
Genetic Factors
Childhood Absences
Although CAE is genetically determined, the precise mode of inheritance and the genes involved remain largely unidentified.9,10,84,154 A positive family history of epilepsy was found in 15% to 44% of cases.20,53,59,62,63,108,127,205 Ascertainment of family history of epilepsy has to be considered: When keeping only epilepsy in parents and siblings, the frequency decreases from 42.6% to 20.7%.111 In two series, epilepsy in first-degree relatives was found in 17% of CAE.36,137 These seizures are TAs and GTCSs. In studies on twins, 84% of monozygotic twins had 3-Hz spike-waves, and TA developed in 75% of pairs and dizygotic twins 16 times less often.127 An Australian study of epilepsy in twins confirmed that in concordant pairs, twins developed a similar syndrome.26 Bianchi et al.28 found that in 24 families with a CAE proband, there was a high concordance (33.3%) for the same clinical form in first-degrees relatives, while febrile convulsions (46.7%) and GTCSs (30%) were more common in distant relatives. Epilepsy risk in children of patients with CAE would be 6.8%.17
Currently, various chromosomal loci have been identified in families with absences of childhood onset (not necessarily equated with CAE). Linkage to chromosome 1 was found in families with absences starting in childhood and the later development of myoclonic jerks and GTCSs, as in JME.58 Linkage analysis in five generations of a family in which affected patients had childhood absences and GTCSs provided evidence of a locus on chromosome 8q24.58,81 The candidate region for this locus, designated ECA 1, has been refined, but a gene remains to be identified. According to the criteria proposed in this chapter, neither of these groups is CAE. There are also reports implicating chromosome 5q31.1 and 19p13.2.52 Furthermore, there is now evidence available to suggest that mutations in genes encoding GABA receptors145 or brain-expressed voltage-dependent calcium channels44 may underlie CAE. Marini et al.145 found GABAA receptor γ-2 subunit gene mutations on chromosome 5 in a large family with CAE and febrile seizures (including febrile seizures plus and other seizure phenotypes). This gene mutation segregated with febrile seizures and CAE, and also occurred in individuals with the other phenotypes. The clinical and molecular data suggested that the GABAA receptor subunit mutation alone could account for the febrile seizure phenotype, but an interaction of this gene with another gene or genes was required for the childhood absence phenotype in this family. Linkage analysis for a putative second gene contributing to the childhood absence phenotype suggested possible loci on chromosomes 10, 13, 14, and 15. Chen et al.44 found 68 variations, including 12 missense mutations in the calcium channel CACNA1H gene in CAE patients. The identified missense mutations occurred in the highly conserved residues of the T-type calcium channel gene.
Juvenile Absences
A family history of epilepsy is frequent, and identical twins who both have the syndrome have been reported.26 Obeid156 reported that in a Saudi Arabian population where consanguineous marriages are frequent and can be found in 47% of the families of patients with JME,152 this was only true for 1 out of 14 JAE families. This could indicate that a recessive gene is important in JME but not JAE. In the clinical genetic study of families with idiopathic generalized epilepsy,146 phenotypic concordance within families of JAE was 10%, which was low compared to families of other IGE syndromes. Because 31% of JAE relatives had CAE but only 2.5% had JME, the authors suggested that CAE and JAE share a close genetic relation, whereas JME may be a more distinct entity. There are several reports of mutation of the CACNA1 A α-1 subunit of the CaV2.1 Ca2+ channel, or the CACNB4 β-4 subunit, with the phenotype of absence seizures with ataxia,72,114 which does not fit in any of the known absence epilepsies. The paper of Escayg et al.72 comprises an observation of a mutation in a family with an identical locus in CACNB4 as in the lethargic mouse mutant but which resulted in a quite different human phenotype. Findings with the genetically well-defined series of mouse mutants with absences can probably not be easily transferred to human epilepsies.
Acquired Factors
As concordance in monozygotic twins is not 100%, nongenetic factors are likely.26 Perinatal complications, postnatal head trauma, and cerebral inflammatory disease were found in the
case histories of 7% to 30% of patients,146,225 and only 3.9% in the Janz series.118 However, these cerebral aggressions are very common in children and were not risk factors in a population-based case-control study.196 A history of febrile convulsions is frequent: 20% to 23% of cases.196,205 More than a risk factor, febrile convulsions are probably the first manifestation of an epileptic diathesis.196 This is in accordance with results of studies on the phenotype of families with “febrile convulsions plus” related or not to mutations of voltage-dependant sodium channel gene mutations (SCN1A or SCN1B). In such families, tonic–clonic, tonic, and absence seizures have been reported.14,128
case histories of 7% to 30% of patients,146,225 and only 3.9% in the Janz series.118 However, these cerebral aggressions are very common in children and were not risk factors in a population-based case-control study.196 A history of febrile convulsions is frequent: 20% to 23% of cases.196,205 More than a risk factor, febrile convulsions are probably the first manifestation of an epileptic diathesis.196 This is in accordance with results of studies on the phenotype of families with “febrile convulsions plus” related or not to mutations of voltage-dependant sodium channel gene mutations (SCN1A or SCN1B). In such families, tonic–clonic, tonic, and absence seizures have been reported.14,128
Pathology and Structural Brain Imaging
Autopsy147,148 and magnetic resonance imaging (MRI)225 studies found microdysgenesis and other cerebral structural changes in some patients with CAE. Meencke and Janz148 reviewed autopsy findings in CAE and confirmed their previous reports on microdysgenesis147 with the frontal lobe more severely affected. Using quantitative MRI, Woermann et al.225 found that patients with idiopathic generalized epilepsy had significantly larger cortical gray matter volumes than control subjects. Abnormalities of the regional distribution of cerebral gray and subcortical matter were frequent in other patients with IGE but only in 1 out of 10 patients with childhood absence epilepsy. However, all cases of Meenke149 had frequent absences from childhood to adulthood and GTCSs, which would not conform to a strict diagnosis of the syndrome of CAE. Similar may be the single patient with abnormal MRI of Woermann et al.225 Recently, thalamic atrophy was demonstrated, using optimized voxel-based morphometry in a series of absence epilepsy patients. Bilateral thalamic atrophy may be either a result of damage from seizures (as in hippocampal sclerosis) in this group of patients with difficult to treat absences or a reflection of a primary underlying pathology as the cause of absence seizures.41
Basic Mechanisms
Current thinking about the pathogenesis of absence seizures dates to the landmark experiments of Jasper and Drooglever-Fortuyn.120 They demonstrated that 3 c/s stimulation of the midline and intralaminar nuclei of the thalamus in cats could produce bilaterally synchronous spike-wave discharges in the cortical EEG of those animals. Over the next 50 years, a debate ensued in the literature as to which was pre-eminent in controlling synchronous spike-wave discharge that characterized absence seizures: The cortex, the thalamus, or both. With the advent of number of animal models of generalized absence seizures, this controversy has been at least partially resolved.210 Moreover, the availability of these models has advanced our understanding of the basic mechanisms of absence seizures. The unifying hypothesis coming from animal data and in vitro neurophysiologic data will be briefly summarized in this chapter.
Animal Models of Generalized Absence Seizures
A valid animal model of generalized absence seizures should reflect the clinical and pharmacologic characteristics of this disorder.42,113,144,155 The criteria for animal models of absence seizures are EEG findings and behavior analogous to human absence epilepsy; reproducibility; predictability; ability to standardize and quantitate; attenuation or blockage by ethosuximide, trimethadione, valproic acid, and benzodiazepines; appropriate ontogeny; exacerbation by γ-aminobutyric acid (GABA)-ergic drugs; blockage by GABAB antagonists; spike-wave discharges that originate in the thalamus, cortex, or both; and hippocampus silent during seizure activity. There are two main genetic models of absence seizures in rats: WAG/Rij46 and GAERS (genetic absence epilepsy rats from Strasbourg).56 A number of other well-characterized genetic mouse models of absence seizures are reported. However, these models manifest a variety of neurologic abnormalities and in some cases other seizures types besides absence epilepsy. Several pharmacologic models of generalized absence seizures are described (γ-hydroxybutyrate [GHB], pentylenetetrazol, penicillin, etc.). These pharmacologic models are all electrographic models because bilaterally synchronous spike-wave discharges are observed.
Unifying Hypothesis
The animal data suggest that three interacting neuropharmacologic forces within the context of the thalamocortical circuitry are involved in the pathogenesis of absence seizures: (a) postsynaptic events required for the occurrence of generalized absence seizures are glutamate-mediated excitatory postsynaptic potentials (EPSPs) followed by GABAA– and GABAB-mediated inhibition that triggers a low-threshold calcium current in nuclear reticularis thalamus neurons; (b) the overall setpoint of thalamic and cortical excitability is modulated by means of ascending cholinergic pathways that project to the thalamus and noradrenergic and dopaminergic neurons projecting to the cortical end of the thalamocortical loop; and (c) presynaptic GABAB and GHB receptors may contribute to the regulation of thalamocortical rhythmicity by means of precise control of excitation and inhibition through modulation of GABA and glutamate release within the involved thalamocortical circuitry.
Genetic Identifiers of Animal Models of Absence Epilepsy
Studies have uncovered the causative genes for absencelike mice models. Tottering and leaner mice have defects in the calcium channel α subunit, lethargic mouse in the calcium channel β-4 gene, and stargazer and waggler mice in the calcium channel γ subunit gene. These mice mutants show some characteristics of absence epilepsy; however, all affected mice show some degree of cerebellar degeneration, which is quite different from human absence epilepsy.110 Attempts to identify the genetic defects in calcium channel genes that underlie human absence epilepsy have so far failed.201 Similar mutations of P/Q-type voltage-gated calcium channel CaV2.1 have been reported in a family with autosomal dominant transmission of absences and episodic ataxia.114 Genetic studies in GAERS suggest a polygenic mode of inheritance of the EEG phenotype with at least three gene loci on chromosomes 4, 7, and 8.197
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


