Chapter 9 Congenital Disorders of the Brain and Spine
TIMING OF FORMATION OF CONGENITAL LESIONS
Myelination
White matter, when myelinated, is bright on a T1-weighted image (T1WI) and dark on a T2WI. The development of myelination is a complicated process but proceeds from posterior to anterior and from central to peripheral (Table 9-1 and Box 9-1). Barkovitch has done a good job in mapping normal myelination dates in term babies. In general, the white matter is seen to be myelinated earlier on T1WI than on T2WI for reasons that have to do with water content versus lipid content. It then takes a bit of wizardry to apply this to the premature infant. Table 9-1 is a good start for determining if there is delayed myelination.
Then along came fluid-attenuated inversion recovery (FLAIR) imaging. The signal intensity of white matter on FLAIR initially is dark because of the marked water content, turns brighter as the “watery brain” of infancy resolves, than again becomes dark as the white matter myelinates. The timing of these stages, summarized in Table 9-1, varies from structure to structure. The cerebellar peduncle and the posterior limb of the internal capsule show high signal intensity relative to gray matter at birth. Thereafter, the white matter loses signal intensity with time and shows low signal intensity at age 50 weeks and beyond. The unmyelinated white matter, including the frontal deep white matter, the occipital deep white matter, and the centrum semiovale, shows low signal intensity at birth, converts to high signal intensity at age 20 to 30 weeks, and back to low signal intensity after age 2 to 3 years.
SUPRATENTORIAL CONGENITAL LESIONS
Arachnoid Cysts
The arachnoid cyst is typically a serendipitous finding and may not be symptomatic. The most common supratentorial locations for an arachnoid cyst are (in decreasing order of frequency) (1) the middle cranial fossa, (2) parasellar cisterns, and (3) the subarachnoid space over the convexities (Fig. 9-1). Infratentorially (see Infratentorial Abnormalities later in this chapter), arachnoid cysts commonly occur in the (1) retrocerebellar cisterns, (2) cerebellopontine angle cistern, and (3) quadrigeminal plate cistern. Intraventricular cysts are rare but favor lateral and third ventricles (Fig. 9-2).
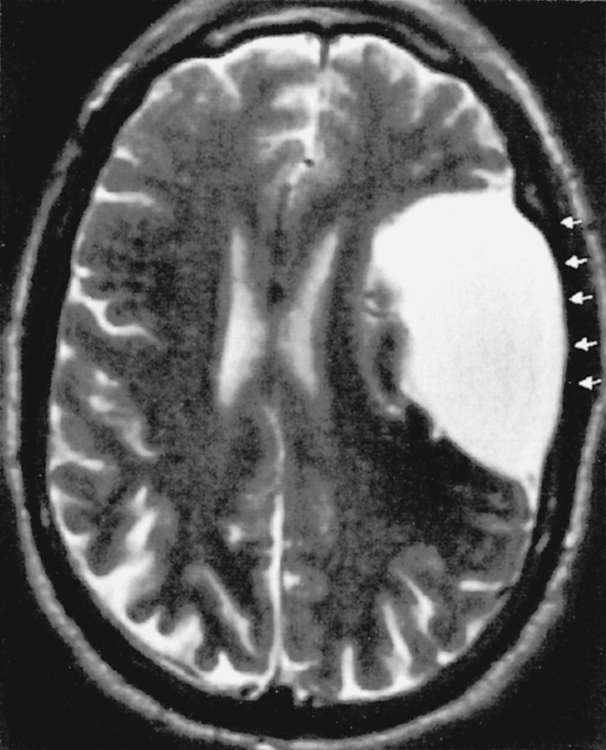
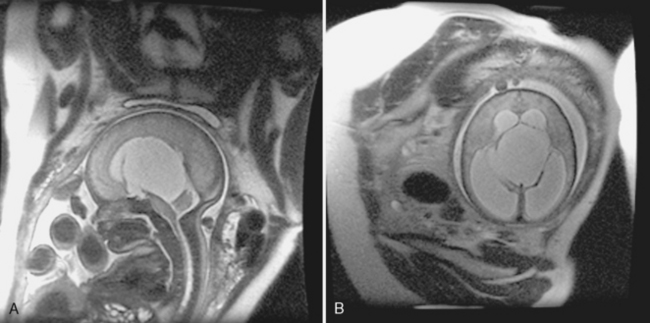
Computed tomography (CT) scans demonstrate a CSF density mass that typically effaces the adjacent sulci and may remodel bone. The mass measures from 0 to 20 Hounsfield units (HU) and shows no enhancement. In those difficult cases where an arachnoid cyst and a dilated subarachnoid space must be distinguished, one can instill intrathecal contrast to differentiate the cyst (which does not immediately fill with contrast) from the subarachnoid space (which immediately fills) (Fig. 9-3). Be careful though. In time, the arachnoid cyst will imbibe contrast.
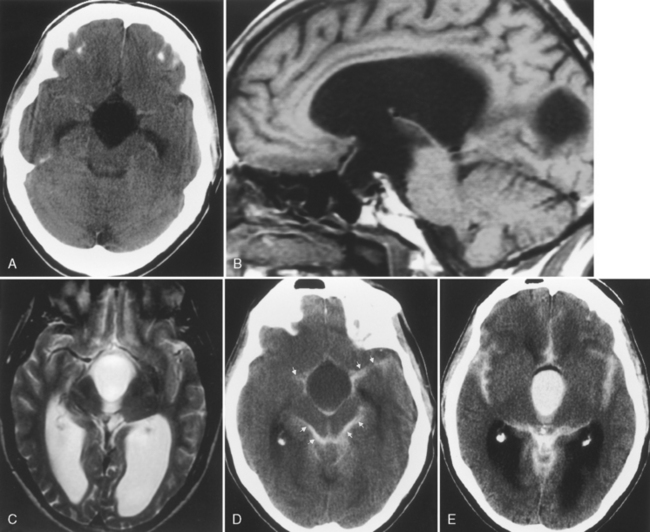
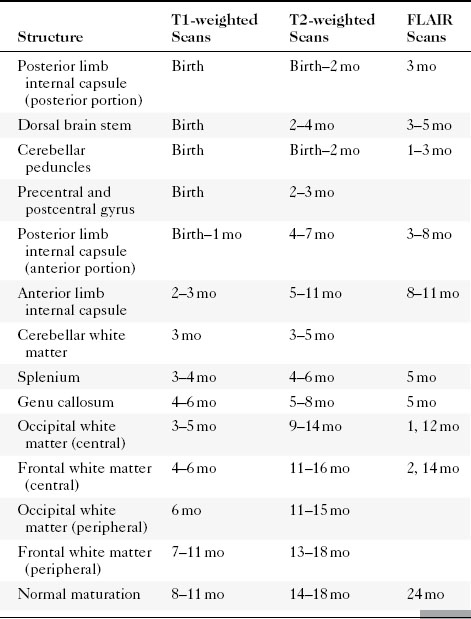
The differential diagnosis of an arachnoid cyst is limited and generally revolves around three other diagnoses: a subdural hygroma, dilation of normal subarachnoid space secondary to underlying atrophy or encephalomalacia, and epidermoid (Table 9-2; see also Fig. 6-1). Although subdural hygromas have been thought to be due to chronic CSF leaks through traumatized leptomeninges, in most cases the trauma results in sufficient blood deposited within the “hygroma” so that the signal intensity on T1WI and FLAIR is different from that of CSF. In addition, subdural hygromas are typically crescentic in shape, whereas arachnoid cysts tend to have convex borders. Both efface sulci and show mass effect. In contradistinction, dilation of the subarachnoid space secondary to underlying encephalomalacia does not demonstrate mass effect and the adjacent sulci are enlarged. Another distinguishing feature is the fact that the cerebral veins in the subarachnoid space are seen to course through the CSF in the case of underlying encephalomalacia, as opposed to the subdural hygroma and arachnoid cyst, where the veins are displaced inward. An epidermoid may simulate an arachnoid cyst on CT and T2WI; however, FLAIR and DWI nicely show higher signal intensity than CSF. In fact, the T1WI will often also be brighter than CSF and will be obvious.
A feature, often seen in association with arachnoid cysts, that may suggest the diagnosis is bony scalloping. The bone may be thinned or remodeled, probably because of transmitted pulsations or slow growth. This would not be seen with hygromatous collections or atrophy. However, the finding may be seen occasionally in epidermoids or with porencephaly, where the ventricular pulsations may be transmitted through the porencephalic cavity to the inner table of the skull (see Fig. 8-21). When arachnoid cysts are seen in the middle cranial fossa, the temporal lobe may be hypoplastic. Absence of soft-tissue intensity or density, calcification, or fat distinguishes arachnoid cysts from those of the dermoid-epidermoid line.
Rathke Cleft Cysts
Rathke cleft cysts are embryologic remnants of Rathke’s pouch, the neuroectoderm that ascends from the oral cavity to the sellar region to form the pituitary anterior lobe and pars intermedia. These cysts are lined with a single layer of cuboidal or columnar cells and may arise within the sella, the suprasellar region, or most commonly both. They occur in up to 13% of autopsy studies of the sella. The cysts can compress normal posterior or anterior pituitary tissue to cause symptoms of hypopituitarism, diabetes insipidus, headache, and visual field deficits but are usually asymptomatic. The cysts are well-defined masses that on MR may have high or low signal intensity on T1WI and high signal intensity on T2WI, and on CT are hypodense and most often nonenhancing (see Fig. 11-31). Intracystic, yellow waxy solid nodules have recently been reported in Rathke cysts containing cholesterol or mucinous proteins probably accounting for the bright signal on T1WI in some cysts. The differential diagnosis is a craniopharyngioma or hemorrhagic pituitary gland; the presence of calcification, soft-tissue mass, or areas of enhancement would lead one away from Rathke cleft cyst as the diagnosis (see Chapter 11). Treatment is cyst aspiration or removal with little chance of recurrence.
Meningoencephaloceles
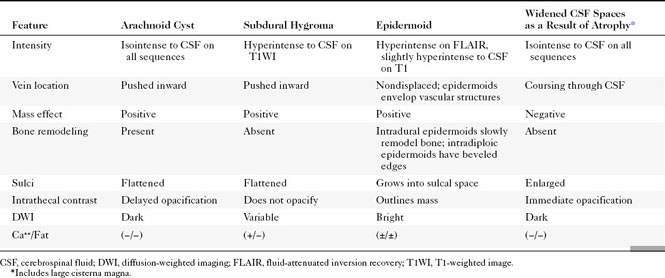
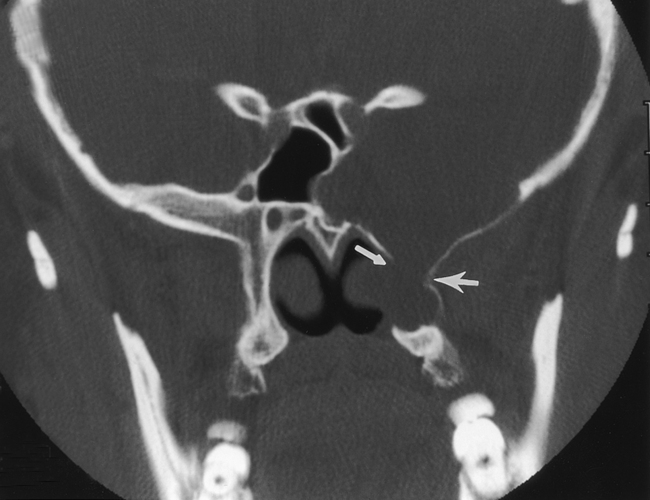
A meningocele is a congenital malformation in which the meninges protrude through the bony confines. As opposed to encephaloceles, meningoceles do not include brain tissue within the protrusion. Usually, an encephalocele also contains meninges, so it should really be called a meningoencephalocele. In fact, clinicians have become resigned to use the shortened noun cephalocele. The terms are used interchangeably in this chapter. Meningoceles are rarer than encephaloceles. The most common location for meningoencephaloceles associated with Chiari III malformations is the occipital region. These may also be associated with holoprosencephaly and aqueductal stenosis (Fig. 9-4). In the United States, parietal (10%) and frontal (9%) meningoencephaloceles are the next most common locations for this abnormality. Vietnamese and Southeast Asian women have a propensity for nasofrontal or sphenoethmoidal meningoencephaloceles (Fig. 9-5). Recent descriptions of meningoceles of Meckel’s cave account for the marked CSF enlargements sometimes seen in these locations. The diagnosis of meningoencephalocele is often made clinically when the protrusion is through the occipital, parietal, or frontal region. On the other hand, nasofrontal or sphenoethmoidal encephaloceles may be clinically occult and may have a wide clinical differential diagnosis when seen through a nasoscope. Otorhinolaryngologists will love you if you let them know that the polyp they plan to remove from the nasal vault is actually the frontal lobe. The anterior meningoencephaloceles have a higher rate of associated anomalies, such as agenesis of the corpus callosum, colobomas, and cleft lips.
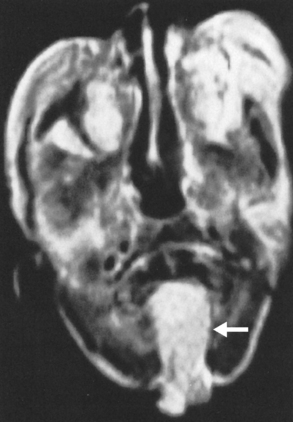
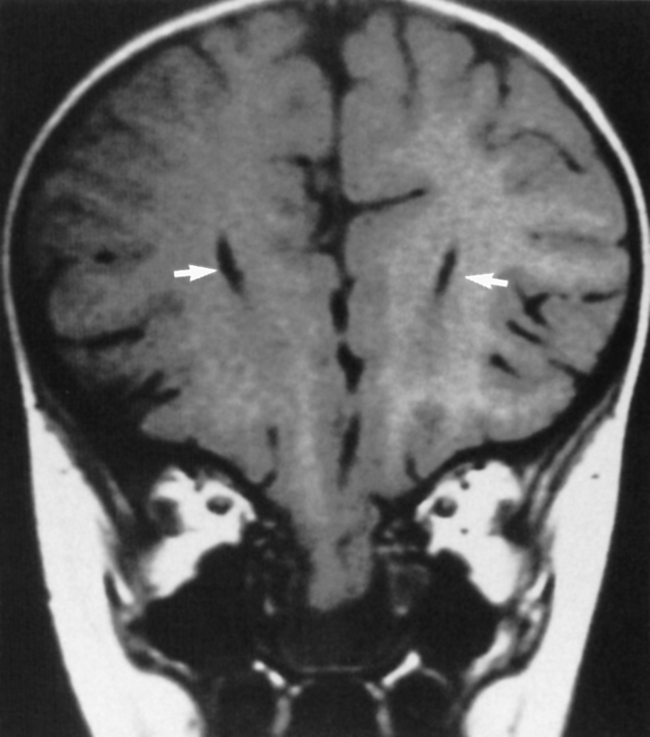
Encephaloceles may also be suspected when one sees enlargement of a basal neural foramen. An MR study will usually discern that there is herniated brain or CSF in the hole (Fig. 9-6). There often is a distortion of the adjacent brain tissue with maloriented white matter tracts.
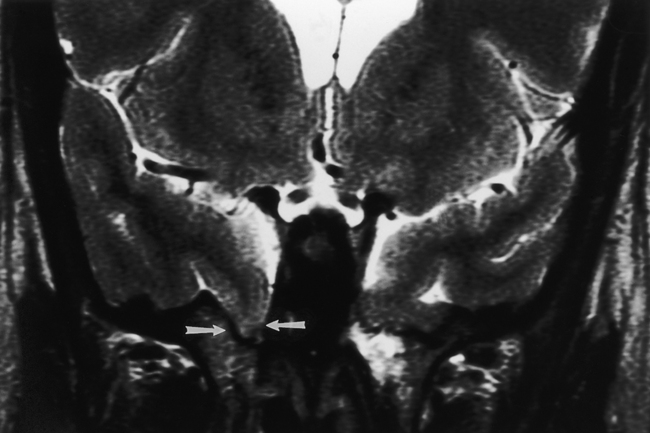
On CT the meningocele is seen as a CSF density protrusion through a bony defect in the calvarium (Fig. 9-7). MR scans demonstrate continuity of the meningocele with the underlying leptomeninges. The critical task is to find the abnormality; secondarily one should tell the surgeons whether brain tissue is protruding through the defect. For this reason MR, with its superior soft-tissue resolution, is the primary study in this evaluation. The brain tissue within the meningoencephalocele typically has the signal intensity of normal brain. However, occasionally one may see heterotopic or disorganized brain tissue, which has a more heterogeneous signal intensity.
Anencephaly
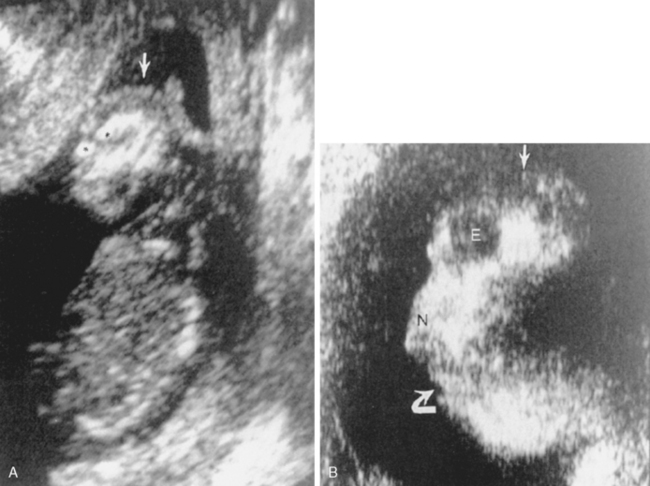
Anencephaly means no brain. Anencephaly should now be a diagnosis made prenatally because most women are screened relatively early in pregnancy for elevated levels of serum α-fetoprotein, a marker of neural tube defects. The diagnosis by obstetric ultrasound (US) is made when the cranial vault is seen to be small, with only the fetus’s face and posterior fossa well seen (Fig. 9-8). Only a nubbin of tissue is seen at the skull base on US, and amniotic fluid α-fetoprotein levels are elevated. These babies do not do well and may be allowed to die if the diagnosis is not made prenatally. An association with spinal dysraphism exists.
Hydranencephaly
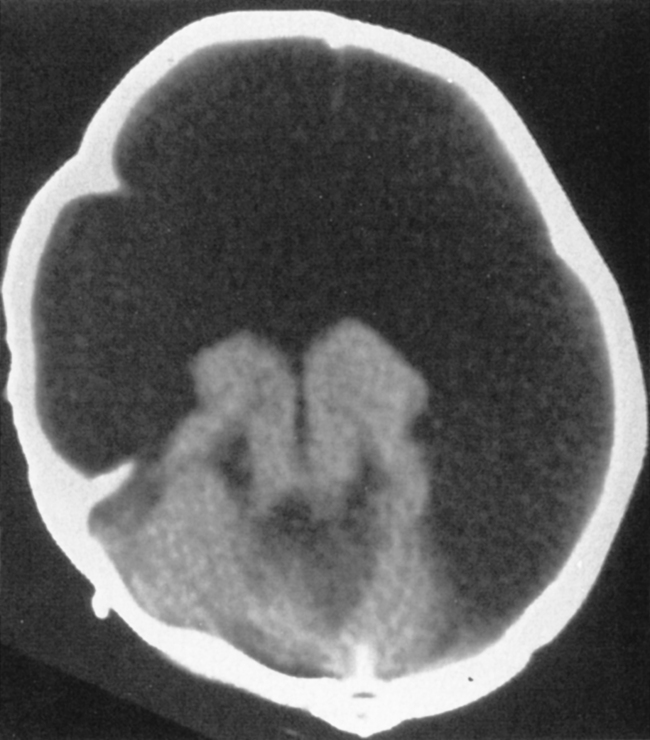
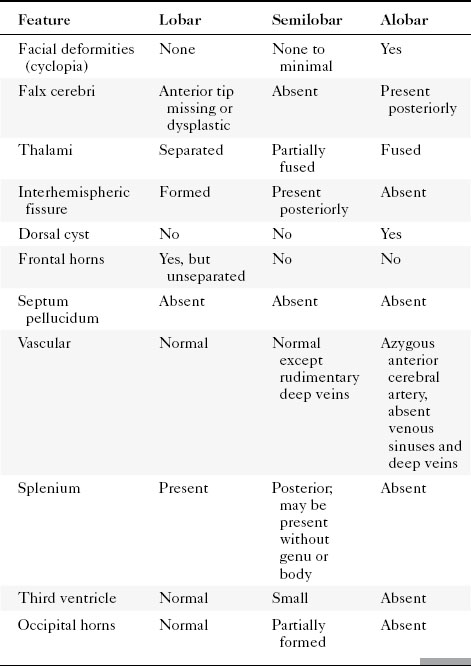
Hydranencephaly has the appearance of absence of that part of the brain supplied by anterior and middle cerebral arteries. The parts of the brain supplied by the posterior circulation are preserved, so the posterior temporal lobes, occipital lobes, thalami, and infratentorial structures are well formed. The brain stops there. In place of the remainder of the brain is a CSF-contained space (Fig. 9-9). The chief differential diagnosis is termed “maximal” hydrocephalus (severe hydrocephalus with a thin cortical mantle plastered against the sides of the calvarium) (Fig. 9-10). This will also usually affect the occipital region as opposed to hydranencephaly. To detect this thin rim of cortical tissue, MR is probably more accurate than CT. Shunting is the treatment for maximal hydrocephalus; some return of function may occur with shunting. With hydranencephaly, no treatment is effective. Bad brain: bad prognosis.
Holoprosencephaly
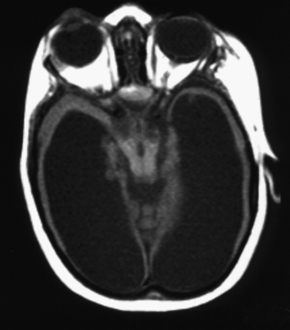
Holoprosencephaly (Table 9-3) refers to a constellation of congenital abnormalities where separation of the right and left cerebral hemispheres is incomplete (Fig. 9-11). The range of this disorder includes a minor form, lobar holoprosencephaly, in which separation of the cerebral hemispheres and lateral ventricles is near normal but formation of an interhemispheric fissure and cerebral falx is incomplete. Even in the mildest of forms you may see fusion of the hypothalamus, cingulum, and caudate nuclei. The frontal horns are closely apposed to each other, with partial fusion of frontal lobes. The most severe form is designated alobar holoprosencephaly, which demonstrates almost no separation of the cerebral hemispheres and ventricles. One is left with a single large ventricle with a horseshoe-shaped appearance (monoventricle) (Fig. 9-12). No interhemispheric fissure, falx, or significant separation of the hemispheric structures is identified. The basal ganglia and thalami are fused, and the septum pellucidum and corpus callosum are absent. Because one needs an interhemispheric fissure to form the corpus callosum, it is not surprising that this is commonly absent in patients with holoprosencephaly. Similarly, a dorsal cyst will interfere with callosal formation in holoprosencephaly. Between the two extremes of lobar and alobar holoprosencephaly is semilobar holoprosencephaly, in which there is partial development of the falx and the interhemispheric fissure (with partial separation of the lateral ventricles) (Fig. 9-13). The basal ganglia and thalami are still fused. These abnormalities are associated with rather severe mental retardation, microencephaly, hypotelorism, and abnormal facies. The finding of a solitary median maxillary central incisor is also indicative of holoprosencephaly.
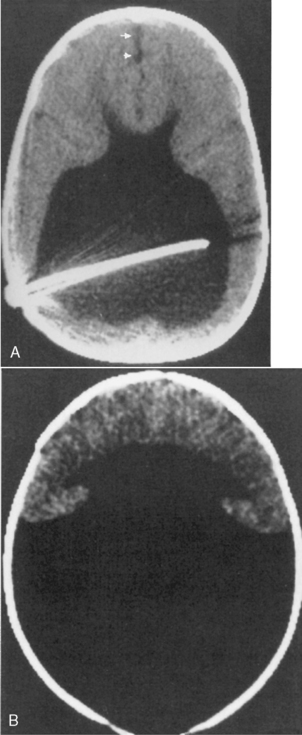
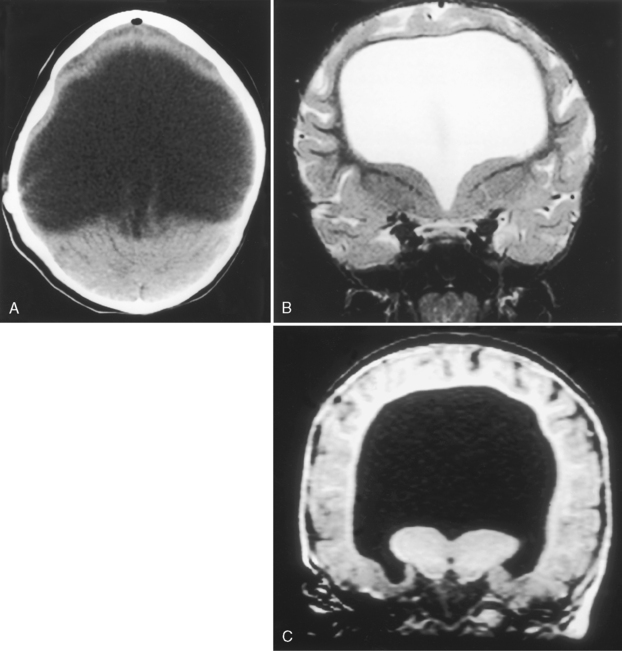
The CT findings in cases of holoprosencephaly include the absence or partial formation of the falx, interhemispheric fissure, and lateral ventricles. Patients typically have absence of the corpus callosum and an abnormally formed third ventricle. MR is a useful first study to examine patients with holoprosencephaly, because the evaluation of the corpus callosum, third ventricle, venous sinuses, vascular abnormalities, and septum pellucidum are better evaluated with MR. The olfactory bulbs and tracts are usually absent with lack of development of olfactory sulci and flat gyri recti. Associations with holoprosencephaly are listed in Box 9-2.
Septo-optic Dysplasia (De Morsier Syndrome)
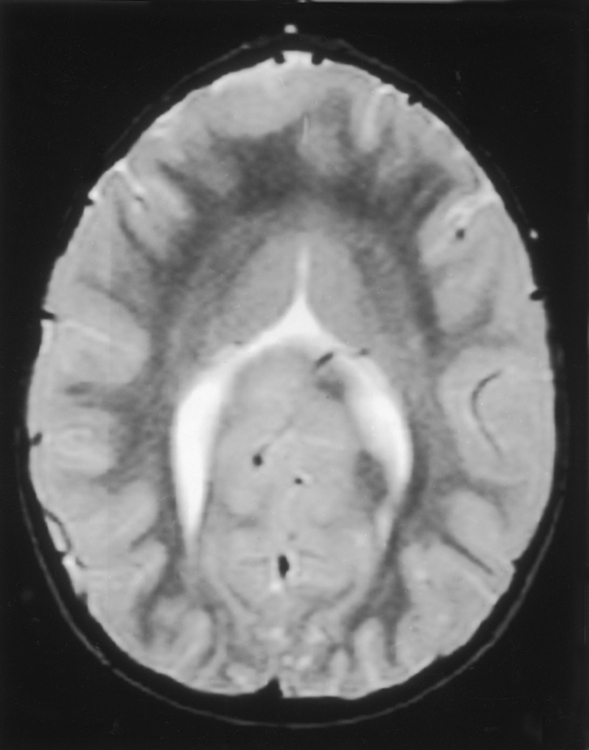
Septo-optic dysplasia is best considered as part of the spectrum of holoprosencephaly. As such, it represents a minor form of holoprosencephaly. The septum pellucidum is either absent (64%) or partially absent (36%), causing a squared-off appearance to the frontal horns of the lateral ventricles (Fig. 9-14). When the septum pellucidum is partially absent, usually the anterior portion is present. This is best seen on coronal MR. Agenesis of the corpus callosum and white matter hypoplasia may be associated with this abnormality. In general, patients with septo-optic dysplasia demonstrate small hypoplastic optic nerves and a small optic chiasm resulting from the dysplastic optic pathways. In some cases that dysplasia may be limited to the optic disk and the nerves/chiasm may not be small. Patients often (60%) have hormonal abnormalities (usually diminished levels of growth hormone) related to the hypothalamic-pituitary axis abnormality. Check for pituitary gland hypoplasia. Seizures coexist. Effects on the visual pathway may range from blindness to normal vision, nystagmus to normal eye movements.
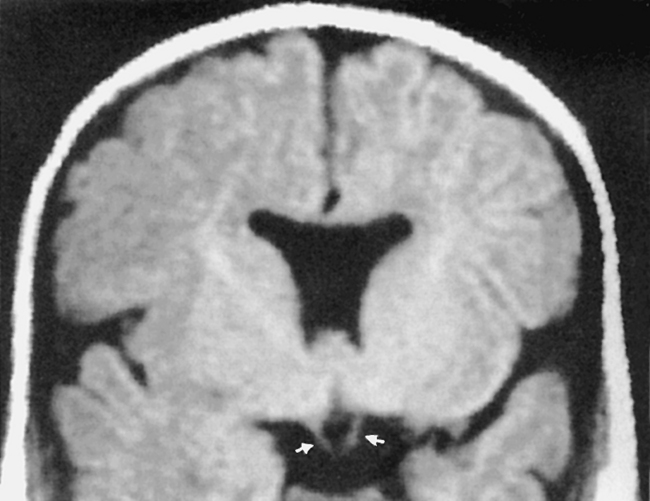
Schizencephaly
Schizencephaly refers to an abnormality of neuronal migration at the fifth to seventh week of gestation in which a cleft is seen coursing from the ventricular ependyma to the pial surface of the brain. This cleft has a gray matter lining (usually from polymicrogyric brain) and is usually seen in the supratentorial space (near the sylvian fissure) coursing to the lateral ventricles. The lips of the cleft may be apposed (closed-lipped) (Fig. 9-15) or gaping (open-lipped). The closed-lipped variety may be missed if the clefts are tightly apposed, but a dimple at the ventricle-cleft interface should suggest the diagnosis. At the ventricular junction there often is associated nodular heterotopia. Schizencephaly occurs most commonly in the frontal (44%), frontoparietal (30%), and occipital (19%) lobes. Bilateral involvement (35% to 67%) is associated with seizures, worse developmental delay, and developmental dysphasia. Motor dysfunction is more common with frontal lobe schizencephaly, open-lipped varieties, and wider gaps in the open lips. Anomalous venous drainage may also be associated with schizencephalic brain. The cause of this disorder is thought to be an ischemic watershed insult in utero that leads to infarction of the germinal matrix and radial glial fibers near the ventricles. If there is failure of the germinal matrix to form, there will be fusion of the pia and the ependymal lining of the ventricles. Normal migration to the cortex is impeded. Other theories espoused as to etiology include toxin exposure, viral infections, and genetic factors. Schizencephaly is often associated with focal cortical dysplasia (polymicrogyria), gray matter heterotopias, agenesis of the septum pellucidum (80% to 90%), and pachygyria. Some believe that focal cortical dysplasias are a form of schizencephaly or vice versa—just that the “cleft” never reaches the ventricle in the former. The lining of the cleft with gray matter, best seen on MR, is the differential point in this lesion and distinguishes it from encephalomalacic abnormalities (porencephaly), which are usually lined by white matter (Fig. 9-16). The inner surface of the cleft is pia-lined and communicates with the ependyma of the ventricle, which differentiates the lesion from an enlarged sylvian fissure seen in premature infants.
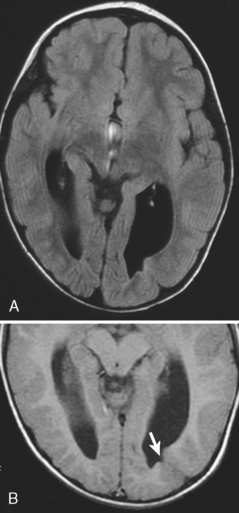
Agenesis of the Corpus Callosum
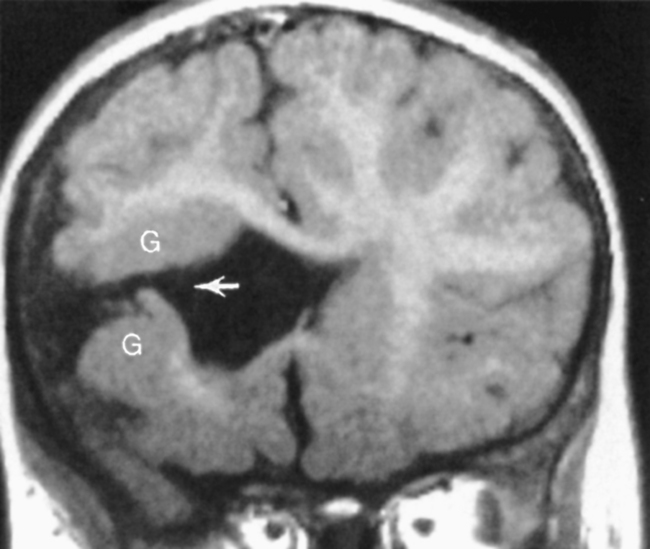
Agenesis of the corpus callosum is one of the more common congenital abnormalities (occurring in 0.7% of all births). Patients with this anomaly often present with refractile seizures or mental retardation. The corpus callosum develops from anterior genu to posterior splenium, accounting for splenial absence in partial agenesis of the corpus callosum. The rostrum is the last portion of the corpus callosum to form, so the combination of absence of the splenium with rostrum agenesis should not put the neoneuroradiologist into a quandary. MR is ideal for the visualization of the corpus callosum because a midline sagittal image demonstrates all the portions of the corpus callosum (Box 9-3). Various degrees of corpus callosum agenesis may occur after complete agenesis; loss of the splenium is the next most common manifestation of this disorder (Fig. 9-17). If the splenium is absent, colpocephaly, dilation of the occipital horns of the lateral ventricles caused by a decrease in the posterior white matter mass, is seen. With complete agenesis, a high-riding, posteriorly oriented, dilated third ventricle; parallel and widely spaced orientation of the lateral ventricles; and impression on the medial aspect of the lateral ventricles because of Probst bundles are seen. The longitudinal Probst bundle is a parallel track of white matter that runs anteroposteriorly and is due to the alternative white matter tract development when the corpus callosum is absent (Fig. 9-18). As the fibers cannot cross, they redirect. The lateral ventricles (especially the occipital horns) are usually moderately dilated and concave medially, as is the third ventricle. Eversion of the cingulate gyrus is also present and the cingulate sulci are therefore not formed. This leads to a picture on the sagittal midline image where the medial hemispheric sulci extend down to the third ventricle. The third ventricle, in turn, extends into the interhemispheric fissure.
Box 9-3 Findings in Agenesis of Corpus Callosum
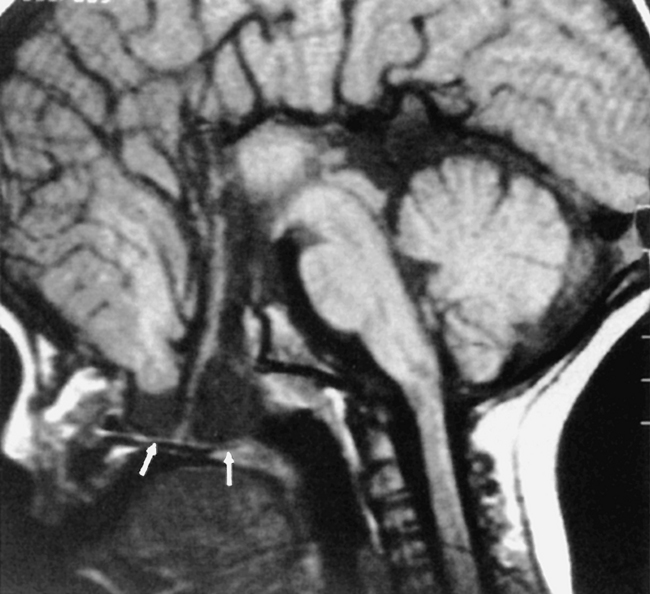
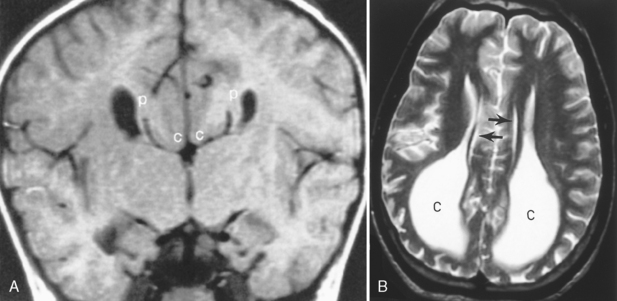
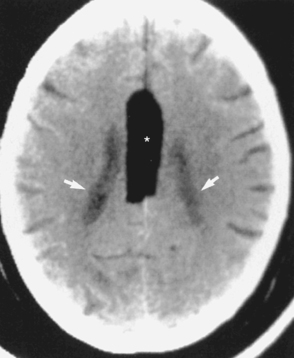
Other midline abnormalities may be associated with agenesis of the corpus callosum (see Box 9-3), such as an interhemispheric arachnoid cyst in the expected location of the corpus callosum or a pericallosal lipoma. A cyst would have low density on CT and signal intensity comparable to CSF on T1WI and T2WI. This can be distinguished from a lipoma (Fig. 9-19), which has fat intensity.
Hamartomas
Hamartomas represent an abnormal proliferation of normal brain tissue in an abnormal location. Whereas the heterotopias are due to anomalous neuronal migration, hamartomas are a nonneoplastic proliferation of well-organized brain tissue. There is a propensity for hamartomatous formation in the hypothalamus (see Fig. 11-39). The patients typically present with precocious puberty; however, occasionally, visual disturbances may be present, because the hypothalamic hamartoma involves the optic pathways. Boys are more commonly affected than girls, and symptoms may also include seizures or laughing spells (gelastic seizures). Because the tissue that makes up a hamartoma is essentially normal brain substance, the hamartoma is isodense with gray matter on CT. On MR, the hamartoma is isointense to gray matter on T1WI and variable on T2WI. The abnormality is identified as a bulbous protrusion of the hypothalamic region in the midline. The hamartomas do not have an abnormal blood-brain barrier and are not expected to show enhancement on either CT or MR. Occasionally, mass effect may be associated with the hamartoma, as evidenced by displacement of the inferior portion of the third ventricle.
ABNORMALITIES OF NEURONAL MIGRATION
Heterotopias
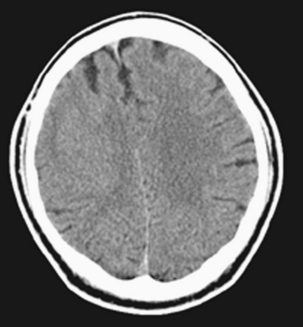
Heterotopic brain is disorganized brain tissue, usually gray matter, that is located in the wrong place because of a premature arrest in neuronal migration from the germinal matrix to the cerebral cortex (Fig. 9-20). Patients usually have seizures, weakness, spasticity, hyperreflexia, or developmental delay. Heterotopias form as a result of an abnormality in neuronal migration at the seventh to sixteenth week of gestation, when migration of the neuroblasts from the periventricular region to the pia is thwarted, possibly because of damage to the radial glial fibers, which orient migrating neurons. The classification of heterotopias is usually divided into two varieties: nodular and band types. Under nodular types, you will find subependymal/periventricular and subcortical variants. A subependymal location is very common, and these gray matter heterotopias are usually truly nodular in appearance (Fig. 9-21) and have the same signal intensity as gray matter. If there is hyperintensity on T1WI, it may be due to dystrophic microcalcifications, and these may show hyperdensity on CT. They may occur as an isolated anomaly or in association with other congenital lesions. A gene, responsible for the protein filamin-1, on the long arm of the X chromosome, has recently been implicated in some cases of nodular subependymal heterotopia. The favored sites are at the trigone or occipital horns of the lateral ventricles. They are bilateral in a high rate, but favor the right side over the left.
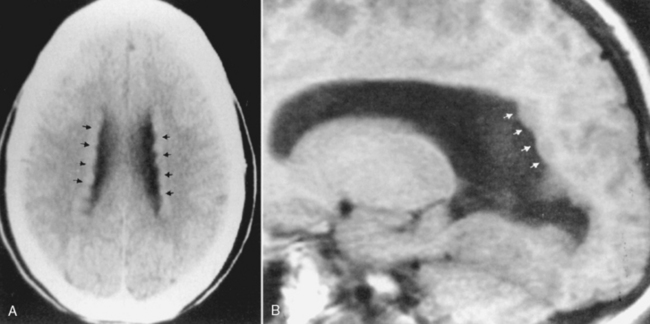
The band type of heterotopias is associated with severe developmental delay and earlier onset of seizures than the focal type (Fig. 9-22). The X-linked gene doublecortin (DCX) predisposes to lissencephaly in men and band heterotopia in women (protected by two X chromosomes). Band heterotopias are isointense with cortical gray matter with well-defined smooth margins. A thin interface of white matter is located between the band (laminar) heterotopia and the cortex, creating the double-cortex sign. The heterotopic tissue may demonstrate mass effect and distort deep gray or white matter structures. The overlying cortex may be abnormal as well due to abnormal sulcation/gyration/migration. Band heterotopias are explained as anomalies arising because of arrest of neuronal migration in the intermediate zone between the germinal matrix and the outer cortex (where the cells belong).
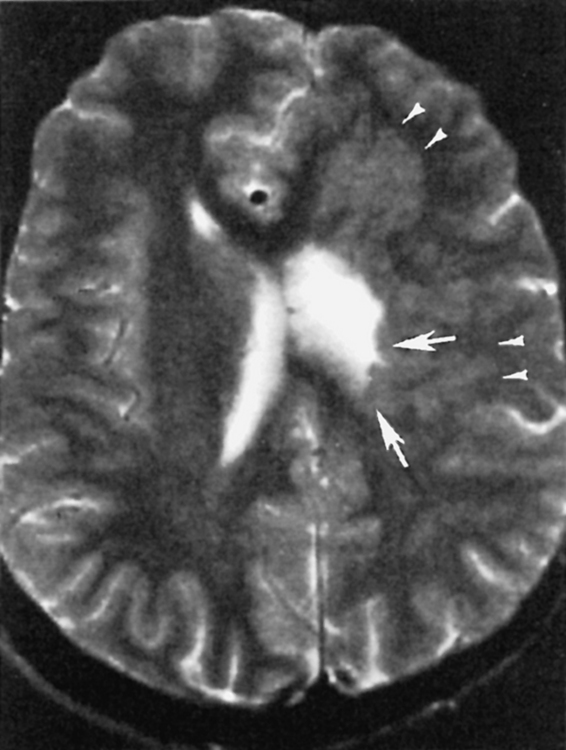
Pachygyria
Pachygyria (incomplete lissencephaly) is a condition of short, broad, fat gyri caused by abnormal sulcation and gyration of the cortical mantle. It is a congenital abnormality that occurs relatively late in gestation, at 12 to 24 weeks, because of neuroblastic migration not proceeding completely to the superficial layers of the cortex (Fig. 9-23). At its most extreme, no sulcation at all may occur, creating a smoothly outlined hourglass configuration to the brain. No sylvian fissures are formed. This condition is termed agyria or complete lissencephaly and is due to abnormal formation of the superficial cortex of the brain, where incomplete neuroblastic migration to the six-layered cortex has occurred, possibly from cortical laminar necrosis of the third cortical layer. A bright rim around the cortex may be seen on T2WI because of this laminar necrosis in the “cell-sparse layer” of the cortex (Fig. 9-24). Patients with congenital cytomegalovirus (CMV) infection have high rates of pachygyria. As opposed to hemimegalencephaly, white matter volume will be decreased; in both, gray matter appears thicker because of poor sulcation. Abnormal myelination may coexist.
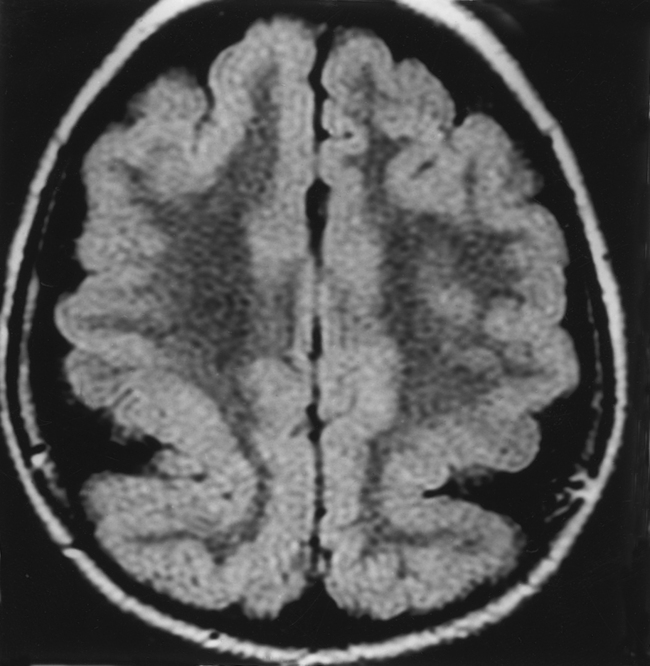
Type 1 Focal Cortical Dysplasia (Without Balloon Cells)
Other MR findings suggesting dysplasia rather than tumor include the presence of homogeneous hyperintense T2WI signal in the subcortical white matter that tapers as it extends to the lateral ventricle (Fig. 9-25). This signal may be due to hypomyelination or wallerian degeneration. Most clinical manifestations are motor, with spastic hemiplegia.
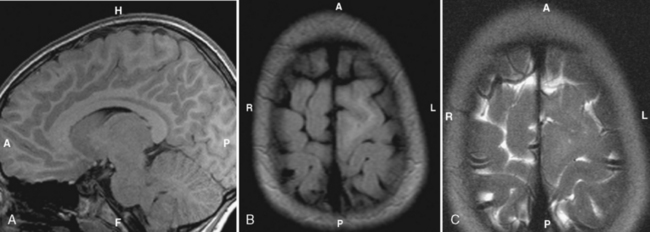
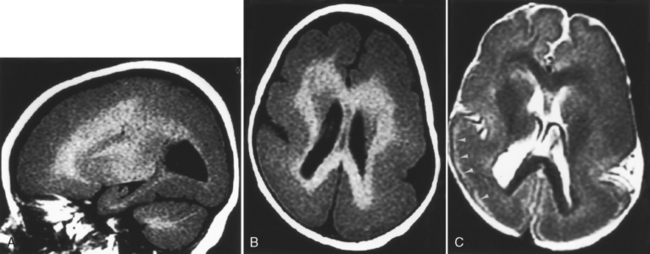
Megalencephaly
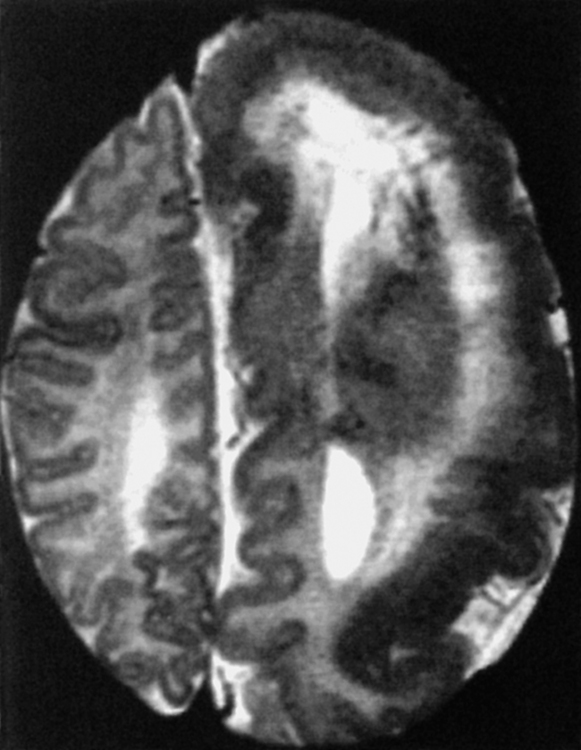
Megalencephaly is defined as enlargement of all or part of the cerebral hemisphere. Bilateral causes (Box 9-4) and unilateral causes (Box 9-5) may have some overlap and account for various head shapes. Often polymicrogyria (associated with increased hemispheric size) or agyria (associated with less severe hemispheric enlargement) is found on the affected side. MR demonstrates a distorted, thickened cortex with ipsilateral ventricular dilation (Fig. 9-26). This unique feature, that of ventricular dilation on the side of the enlarged hemisphere, separates congenital hemimegalencephaly from other infiltrative lesions. This disorder results from too many neurons and decreased apoptosis. Heterotopias and cortical thickening are characteristic. Myelination is delayed, but overall white matter volume is increased. Alternatively, megalencephaly may be an isolated finding. Patients have seizures, hemiplegia, developmental delay, and abnormal skull configurations.
Lipomas
Lipomas have attenuation values on CT that are in the negative range, usually −30 to −100 HU, and are isodense to subcutaneous fat (see Fig. 9-19). High in intensity on T1WI, intermediate to low in intensity on conventional T2WI, and bright on fast spin-echo T2WI, the fat of the lipoma can be corroborated by the presence of a chemical shift artifact along the frequency-encoding direction. This causes one edge of the lesion to be highlighted as very bright on T2WI and the other edge along the frequency-encoded axis to be darkened. Alternatively, a fat-suppression pulse sequence can verify the fat by demonstrating signal diminution after it is applied. Calcification may be seen.
Aqueductal Stenosis
Aqueductal stenosis, causing lateral and third ventricular enlargement without fourth ventricular dilation, is usually seen in boys as part of an X-linked recessive hereditary disorder. This is thought to be due to an abnormality of proliferation and differentiation of the periaqueductal gray matter of the mesencephalon, leading to the stenosis. Acquired causes of aqueductal stenosis are numerous and include clots from subarachnoid hemorrhage and fibrosis after bleeds or infections. The congenital causes occur in the setting of enlarged head circumference in infant boys and may be due to webs, septa, or membranes. This entity is covered in greater depth in Chapter 8.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


