Congenital Intracranial Malformations
Congenital malformations of the brain comprise a large and diverse set of anomalies that result in a wide range of functional outcomes for affected children—ranging from relatively benign neurologic deficits to severe deficits that are incompatible with life. The nervous system develops in an orchestrated manner, regulated by numerous proteins that signal the proliferation, migration, and differentiation of primordial cells into the functioning neurons and supporting structures that make up the human nervous system. Cerebral malformations, thus, can result from dysfunction along this highly regulated embryogenic pathway through genetic, metabolic, infectious, and in utero environmental abnormalities. Advances in neuroimaging make possible the identification of more subtle malformations. In addition, the increasing availability of high-resolution microarray-based comparative genomic hybridization (aCGH) genetic testing has allowed a more precise diagnosis than ever before. In fact, the explosion in our knowledge of the genetic and molecular controls involved in brain development is shedding new light on the etiologies of congenital malformations, raising the possibility of improving our management of these often devastating conditions.
Based on the current understanding of abnormal embryology, the major malformations are grouped as supratentorial or infratentorial (see boxes “▶ Classification of Supratentorial Malformations” and “▶ Classification of Infratentorial Malformations”). As we continue to gain more knowledge of the developing brain in the future, the classification will likely be altered. A detailed description of the development of the central nervous system (CNS) is covered in another chapter. We discuss embryology only briefly here as it pertains to the various congenital malformations. The chapter concludes with some general comments and recommendations regarding their diagnosis and surgical management.
Classification of Supratentorial Malformations
Disorders of neurulation
Anencephaly, craniorachischisis
Cephalocele (encephalocele)
Congenital dermal sinus
Disorders of ventral induction
Holoprosencephaly
Septo-optic dysplasia
Dysgenesis of the corpus callosum
Abnormal neuronal proliferation or apoptosis
Reduced proliferation or excess apoptosis
Microcephaly
Microlissencephaly
Megalencephaly
Cortical dysgenesis with abnormal cell proliferation
Hemimegalencephaly
Type 2 focal cortical dysplasia
Congenital neoplasia
Abnormal neuronal migration
Neuroependymal abnormality
Periventricular (subependymal) heterotopia
Abnormal transmantle migration
Lissencephaly/subcortical band heterotopia
Abnormal late transmantle migration
Subcortical heterotopia
Abnormal terminal migration
Cobblestone complex
Abnormal postmigrational development
Polymicrogyria
Polymicrogyria with schizencephaly
Polymicrogyria without schizencephaly
Focal cortical dysplasia
Type 1 focal cortical dysplasia
Type 2 focal cortical dysplasia
Microcephaly
Classification of Infratentorial Malformations
Malformations of the midbrain and hindbrain
Molar tooth sign malformations
Joubert syndrome
Joubert syndrome–related disorders
Rhombencephalosynapsis
Malformation of the cerebellum
Cerebellar dysplasia
Lhermitte-Duclos disease
Migration disorders
Vermis and cerebellar hemisphere hypoplasia
Focal
Diffuse (e.g., Dandy-Walker malformation)
Cerebellar vermis hypoplasia
Isolated
Syndromic
Malformation of the pons and cerebellum
Pontocerebellar hypoplasia
Data from Parisi and Dobyns.136
15.1 Types of Congenital Intracranial Malformations
15.1.1 Anencephaly
Anencephaly is the most severe of the neural tube defects (NTDs), resulting in the absence of major portions of the brain and skull. Like craniorachischisis and spina bifida, anencephaly occurs when the rostral neural tube fails to close. Apposition and fusion of the neural apices at the dorsal midline occur between the third and fourth weeks of gestation. The fusion begins at the primitive hindbrain–cervical junction, referred to as closure 1, and spreads in both a rostral and a caudal direction. Additional points of fusion occur at the most cephalic end of the embryo, known as closure 3, and at a variable location between closures 1 and 3, known as closure 2.1,2 A defect in fusion of the anterior neuropore, between closures 1 and 3, at approximately gestational day 24, results in anencephaly (▶ Table 15.1).3 Initially, a rudimentary cerebrum can exist, resulting in what is known as anencephaly (▶ Fig. 15.1), but the exposed brain is subject to injury from amniotic fluid and in utero mechanical trauma. As a result, stillbirths are common with anencephaly, but a third of affected fetuses are born alive. Unfortunately, there is no effective treatment, and these newborns survive only hours. They are thought to be not capable of consciousness or of experiencing pain owing to the lack of cerebral development.4
| Gestational age | Normal development | Malformation | Genes |
| Development of the hemispheres | |||
| 3–4 weeks | Primary neurulation | Anencephaly Cephalocele Congenital dermal sinus | |
| 6 weeks | Prosencephalic cleavage | Holoprosencephaly | SHH, SIX3, TGIF, ZIC2, PTCH, BMP, HPE1, HPE6, HPE8 |
| 6–12 weeks | Midline prosencephalic development | Septo-optic dysplasia | HESX1, SOX2, SOX3, OTX2 |
| 7–20 weeks | Development of corpus callosum | Dysgenesis of corpus callosum | |
| 2–4 months | Neuronal proliferation and differentiation | Microcephaly Megalencephaly Hemimegalencephaly Focal cortical dysplasia, type 2 Congenital neoplasias | MCPH1, ASPM, SLC25A19, TSC1 |
| 3–5 months | Neuronal migration | Lissencephaly/subcortical band heterotopia Cobblestone complex Neuronal heterotopias | LIS1, DCX (XLIS), ARX, RELN TUBA1A FCMD, FKRP, LARGE, POMGnT1 FCMD (Fukutin), MEB, POMT1 FLNA (FLN1) |
| gestational month 5 to postnatal years | Cortical organization | Polymicrogyria Focal cortical dysplasia, types 1 and 3 Schizencephaly | GPR56, WDR62, SRPX2, PAX6 EMX2 |
| Development of the brainstem and cerebellum | |||
| 4–5 weeks | Midbrain–hindbrain patterning | Joubert syndrome and related disorders | |
| gestational week 5–postnatal month 7 | Rhombic lip development and neuronal proliferation | Rhombencephalosynapsis Cerebellar hypoplasias Pontocerebellar hypoplasias | TSEN54, RARS2, CLAM, VRK1 |
| gestational month 2–postnatal month 20 | Migration and differentiation of cerebellar neurons | Lhermitte-Duclos disease | PTEN |
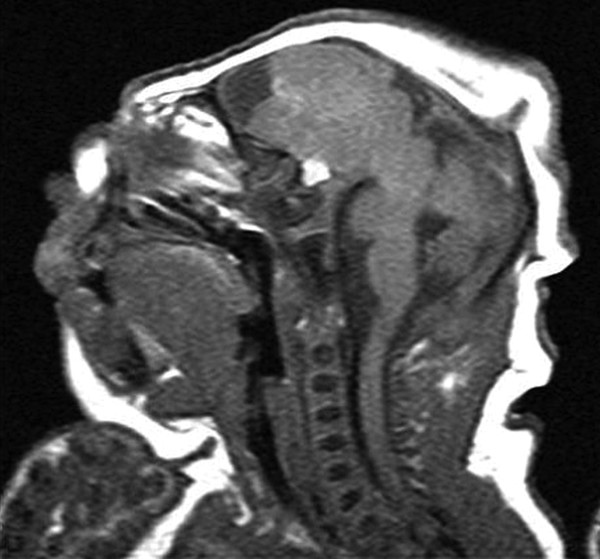
Fig. 15.1 Sagittal T1-weighted magnetic resonance image of a neonate born with anencephaly. All of the occipital and parietal lobes and most of the frontal lobes are degenerated. The posterior fossa structures are nearly intact. Note the proptotic eye and recessed forehead.
The diagnosis of anencephaly can be made prenatally by screening alpha fetoprotein and ultrasound. Currently, anencephaly occurrs an estimated 1 in 1,000 births in the United States,5,6 although there are significant geographic, racial, and socioeconomic variations. However, because of the option of pregnancy termination after prenatal diagnosis, the exact incidence is difficult to determine. The introduction of maternal folic acid supplementation has resulted in a significant decline of NTDs worldwide, including anencephaly.
15.1.2 Cephalocele
A cephalocele (or encephalocele) is a rare form of NTD in which the cranial contents protrude through a congenital defect in the cranium. It can be further classified based on the contents. A cranial meningocele contains only meninges, whereas an encephalomeningocele or encephalocele contains both meninges and brain tissue (▶ Fig. 15.2). An encephalocystocele or hydrencephalomeningocele also includes a dilated ventricle. These malformations are a result of failure of the neuroectoderm to separate from the surface ectoderm during neural tube closure, which leads to a mesodermal defect and prevents proper ossification of the skull. If the skull defect is sizeable, meninges and brain can herniate through the opening, resulting in a meningocele or cephalocele. The sac, which is often at the midline, is covered by normal or dysplastic skin, and thus, maternal serum alpha fetoprotein is typically normal. Cephaloceles in the occiput are most often seen in North America, and those in the frontal region, particularly the nasofrontal, nasoethmoidal, or naso-orbital region, are common in Southeast Asia, certain parts of India, and Africa.7,8 The prognosis depends on the location of the lesion, the presence and size of brain herniation, and other congenital anomalies. Cephaloceles can be associated with cerebellar dysplasia, lissencephaly, hypoplastic brainstem, and Dandy-Walker malformations, as well as cleft lip, cleft palate, hypertelorism, microphthalmia, anophthalmia, syndactyly, and heart defects. Surgical options must be discussed in this context.
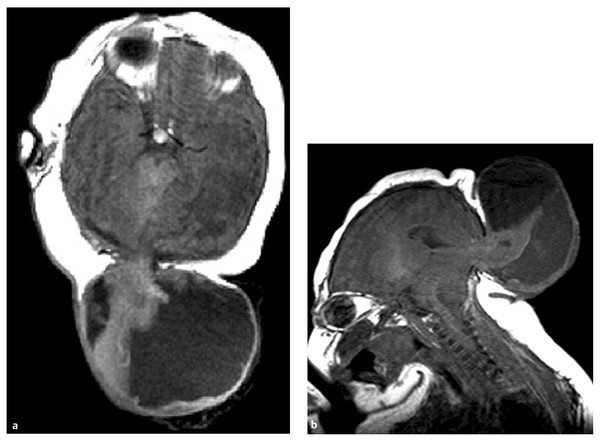
Fig. 15.2 Axial T1-weighted (a) and sagittal T1 weighted (b) magnetic resonance images of a newborn with occipital cephalocele show suboccipital cranial defect and herniated cerebellum.
15.1.3 Congenital Dermal Sinus
A dermal sinus is a tract lined by keratinized squamous epithelium originating from a skin dimple with variable intracranial or intraspinal extension. It results from a focal adhesion and incomplete dissolution of the continuity between the neural and cutaneous ectoderm during neurulation, around the fourth week of gestation.9,10 The persistent tract can occur anywhere between the columella of the nose and the coccyx. Nearly all reported cranial cases occur in the midline and most commonly involve the external occipital protuberance (85%) and less often the nasion (10%) and the posterior parietal area (5%). Inclusion cysts (dermoid cyst, epidermoid cyst, and teratoma) may exist at any point along the tract.11–13 Dermal sinuses can connect the skin surface to any depth within the CNS and thus can be a source of intracranial infections and should be excised.14
15.1.4 Holoprosencephaly
Holoprosencephaly (HPE) is the most common developmental disorder of the human forebrain and is the result of failure of the forebrain to divide into two distinct hemispheres. Although it occurs once in every 250 conceptuses, most of these do not survive to term, and HPE is seen in only 1 in 10,000 live births and stillbirths.15,16 Few such newborns survive past 1 year.17 HPE appears to be more common in girls, nonwhite races, and multiple-gestation pregnancies. Newborns with HPE often have a low birth weight. Genetic and environmental causes of HPE are thought to disrupt the prechordal plate during early gastrulation and the neural plate during neurulation.15,18 Perturbation of the prechordal mesoderm disrupts neural induction and regional patterning, both of which are critical to normal forebrain and midface development. Nonsyndromic, nongenetic causes are not common, and implicated teratogens include maternal diabetes, ethanol, antiepileptic drugs, retinoic acid, cigarette smoking, congenital cytomegalovirus infection, and alkaloids of the plant Veratrum californicum.19–21
More than a third of patients with HPE have chromosomal number abnormalities, particularly trisomy 13, followed by trisomy 18 and triploidy, as well as certain malformation syndromes, such as Smith-Lemli-Opitz syndrome.22,23 Transmission can be autosomal-dominant, autosomal-recessive, or X-linked. The primary pathway implicated in HPE and other midline defects is the Sonic Hedgehog (SHH) signaling pathway.24 Inheritance of HPE exhibits an 80% penetrance, most commonly related to several genes, including SHH on 7q36, ZIC2 on 13q32, SIX3 on 2p21, and TGIF on 18p11.3.24 Less common genes implicated include GLI2 on 2q14 and NODAL on 10q22.11. Disrupted gradients of gene expression along the three anatomical axes are thought to be elementary in the pathogenesis of HPE. Variations in the timing and location of gene mutations help to account for the diverse phenotypic spectrum in those affected by HPE. The genetics of HPE is thought to be a complex, multiple-hit process because a genetic mutation cannot be identified in up to 83% of HPE cases, although the increasing use of aCGH is beginning to change the landscape.15,24
The classification put forth by DeMyer and colleagues in the 1960s still serves as a useful starting point in understanding this heterogeneous disease.25 The most severe form, alobar HPE, is characterized by an almost complete lack of midline cleavage between the hemispheres, resulting in the classic “monoventricle” of the forebrain (▶ Fig. 15.3). This single ventricle is often associated with a dorsal cyst, and there is no identifiable corpus callosum or interhemispheric fissure. Semilobar HPE forms when there is some separation of the posterior hemispheres and occipital horns but the anterior hemispheres and associated ventricles fail to separate. Most of the corpus callosum fails to develop, except for the splenium. The least severe form of classic HPE is the lobar type, in which the lack of midline cleavage is restricted to the rostral–ventral forebrain with near-normal separation of the remainder of the forebrain. The corpus callosum is normal with the exception of the genu. A more recently recognized and milder subtype of HPE, known as the middle interhemispheric variant, results from the failed separation of the posterior frontal and parietal lobes but spares the rostral–ventral frontal area.17,26
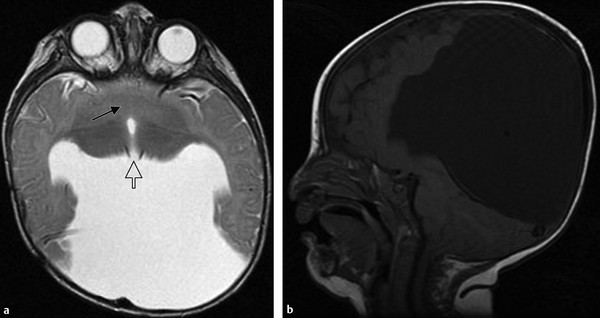
Fig. 15.3 Axial T2-weighted (a) and sagittal T1-weighted (b) magnetic resonance images of a child with alobar holoprosencephaly show fused basal ganglia (arrow) and a monoventricle that opens to a dorsal cyst. The thalami are partially fused (open arrow).
Superimposed on the different forms of HPE described above is variability in the cleavage of the deep gray nuclei.27 In every case of HPE, there is some degree of hypothalamic noncleavage. The next most likely deep gray structures not to cleave are the caudate nuclei, followed by the lentiform nuclei and finally the thalami, which remain fused in 67% of patients. The extent of fusion of these deep gray nuclei, particularly the basal ganglia and thalami, is proportional to the hemispheric noncleavage described by DeMyer and has prognostic value for neurologic outcome.28 However, there are noted exceptions to this trend, which may in part account for the variation in the severity of neurologic dysfunction among patients with the same type of HPE under DeMyer’s classification. The variability of morphologic nonseparation and phenotypic expression of HPE precludes a simple and accurate prognosis for each patient.
It has long been observed that the “face predicts the brain.” In other words, the various forms of midface hypoplasia often associated with HPE serve as markers of the underlying forebrain abnormalities.29 Severe midface abnormalities, such as cyclopia, ethmocephaly, and cebocephaly, often coincide with alobar HPE.18,30 Other associated midline abnormalities can be relatively subtle and include arhinencephalia, agenesis of the corpus callosum, pituitary abnormalities, median cleft lip, and the presence of a single maxillary central incisor.31 Clinically, patients can present with oral motor dysfunction, seizures, endocrinopathies (especially diabetes insipidus), microcephaly, hydrocephalus, dystonia, spasticity, choreoathetosis, and developmental delay. The hydrocephalus and third ventricular dorsal cyst of alobar HPE are related to blockage of the outlet of the third ventricle in conjunction with aqueductal stenosis.22
15.1.5 Hydranencephaly
Hydranencephaly results from destruction of a previously normally developing brain by in utero vascular occlusions and hemorrhages, infection, or maternal exposure to drugs, cigarette smoking, or radiation.32 The hemispheres corresponding to the middle cerebral artery or, more often, the entire anterior circulation is resorbed and replaced with cerebrospinal fluid (▶ Fig. 15.4). The thalamus, brainstem, and cerebellum are spared along with the meningeal sac and falx. There may be a small residual occipital, frontal, or temporal cortex present. Hydranencephaly, which appears on imaging as nearly complete absence of the cerebral hemispheres, can be difficult to differentiate from HPE with hydrocephalus. The presence of a falx and separate thalami, along with the absence of facial anomalies and the frontal cortical mantle, distinguishes hydranencephaly from HPE.
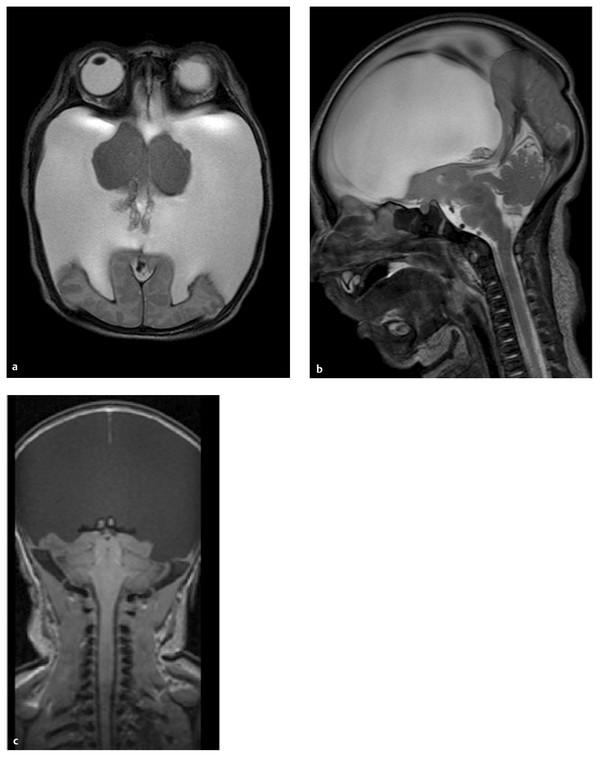
Fig. 15.4 Gradient-echo axial (a) and sagittal T2-weighted (b) and coronal T1-weighted (c) magnetic resonance images of child with hydranencephaly. The cerebral hemispheres, except for a thin rim of the occipital cortex, are absent and replaced by cerebrospinal fluid. The thalamus, brainstem, and cerebellum are preserved. Unlike in HPE, the falx in fully formed.
15.1.6 Septo-optic Dysplasia
Septo-optic dysplasia (SOD), named by de Morsier in 1956, is a disorder defined by any combination of the classic triad of optic nerve hypoplasia, pituitary dysfunction, and midline brain defects, particularly absence of the septum pellucidum (▶ Fig. 15.5). There is considerable variation in the phenotype, with fewer than a third of patients having all of the manifestations33; however, all patients have optic nerve hypoplasia, although the degree of visual impairment and nystagmus is variable. Approximately one-half to two-thirds of patients have an endocrinopathy. Of these, almost all have dysplasia of the hypothalamic–pituitary axis, including a truncated or absent pituitary stalk, a hypoplastic anterior pituitary lobe, and an ectopic or absent posterior pituitary gland.34 Pituitary hormone insufficiency is more frequent in those missing a septum pellucidum than in those with an intact septum. Most patients with SOD are of normal intelligence, but those with developmental delay often have additional brain malformations, including cortical dysplasia and schizencephaly, and they are diagnosed as having SOD+.35,36 However, when schizencephaly or polymicrogyria is present, the child typically has minimal or no pituitary dysfunction.37
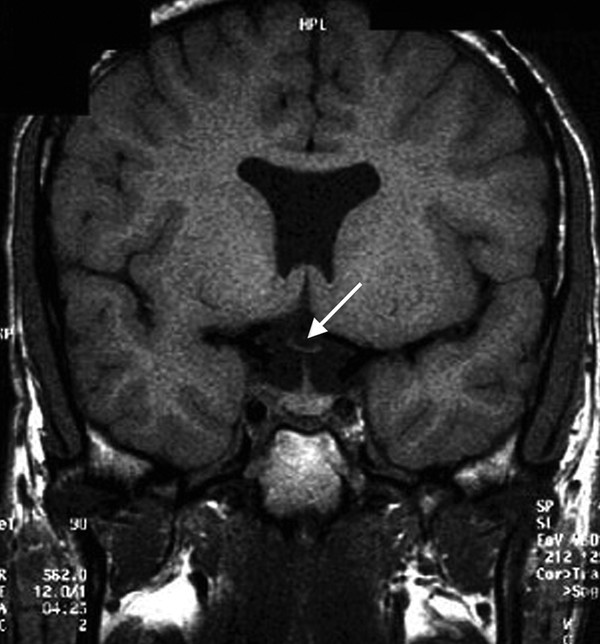
Fig. 15.5 Coronal T1-weighted magnetic resonance image of septo-optic dysplasia shows absence of the septum pellucidum and a hypoplastic optic chiasm (arrow). The frontal horns of the lateral ventricles have a characteristic horizontal roof.
The management of children with SOD includes the early detection and replacement of deficient pituitary hormones. Although the etiology of SOD remains unclear, maternal diabetes mellitus, cytomegalovirus infection, and exposure to toxins and mutations in four key genes have been implicated: HESX1, SOX2, SOX3, and OTX2. Abnormalities in the interplay between these key regulators at the prechiasmatic plate during development determine, in part, the severity of midline brain maldevelopment.38
15.1.7 Dysgenesis of the Corpus Callosum
Agenesis of the corpus callosum (ACC), like SOD and HPE, is a disorder of midline development. It occurs commonly, affecting 0.7% of the general population and 2.3% of the developmentally disabled population.39,40 It has a prevalence of between 1 in 4,000 and 1 in 5,000 live births.41 It can occur in isolation or concurrently with other CNS malformations and is a feature in more than 20 malformation syndromes (see box “▶ Disorders Associated with Agenesis of the Corpus Callosum”). In all circumstances, the common pathogenesis is the failure of cortical cells to grow axons across the midline. Failure to develop the dorsal portion of the rostral wall of the midline prosencephalon starting in the middle of the second month of gestation prevents the formation of the commissural plate through which callosal fibers cross the midline. Axons that would normally form the corpus callosum are diverted, forming longitudinal bundles of Probst in a location superior and medial to the lateral ventricles (see box “▶ Magnetic Resonance Imaging Findings of Agenesis of the Corpus Callosum”).42 Disruption of the migration and differentiation of callosal neurons, impairment of the molecular control of axon growth (e.g., DCC and L1 adhesion molecule), or loss of the guidance of axons traversing the midline by chemoattractants (e.g., netrin 1 and Slit protein) also lead to ACC, but Probst bundles are characteristically absent.43 This often occurs in lissencephaly with ACC (e.g., Walker-Warburg syndrome).
Disorders Associated with Agenesis of the Corpus Callosum
Always present
Aicardi syndrome
Andermann syndrome
L1 disease
Mowat-Wilson syndrome
Chiari type 2 malformation
Occasionally present
Holoprosencephaly
Septo-optic dysplasia
Dandy-Walker malformation
Lissencephaly (Walker-Warburg syndrome, Miller-Dieker syndrome)
Other disorders of neuronal migration
Fetal alcohol syndrome
Metabolic disorders
Trisomies 8, 13, and 18
Magnetic Resonance Imaging Findings of Agenesis of the Corpus Callosum
Separation and parallel orientation of the lateral ventricles
Radial orientation of the interhemispheric gyri
Colpocephaly (dilated occipital ventricular horns)
Probst bundles
Inversion of the hippocampi
Absent septum pellucidum
Rostral extension of the third ventricle
Hypoplastic or enlarged anterior commissure
Azygous or wandering anterior cerebral artery
Absent cingulate sulci with everted cingulate gyri
In partial ACC, the commissural plate is formed, but an arrest of the developing commissure between weeks 7 and 20 of gestation results in an incomplete corpus callosum44 (▶ Fig. 15.6). Typically, the posterior body, splenium, inferior genu, and rostrum of the corpus callosum are missing because they are thought to develop last. However, dysgenesis of the anterior body, genu, and rostrum with a normal splenium has been observed, particularly with semilobar HPE.45 This finding suggests that the growth of the corpus callosum is bidirectional instead of anterior to posterior, as has been widely accepted in the past. Lipomas, arising from the remnant of the meninx primitiva, can also prevent full growth of the corpus callosum by mechanical obstruction of the crossing axons.46,47
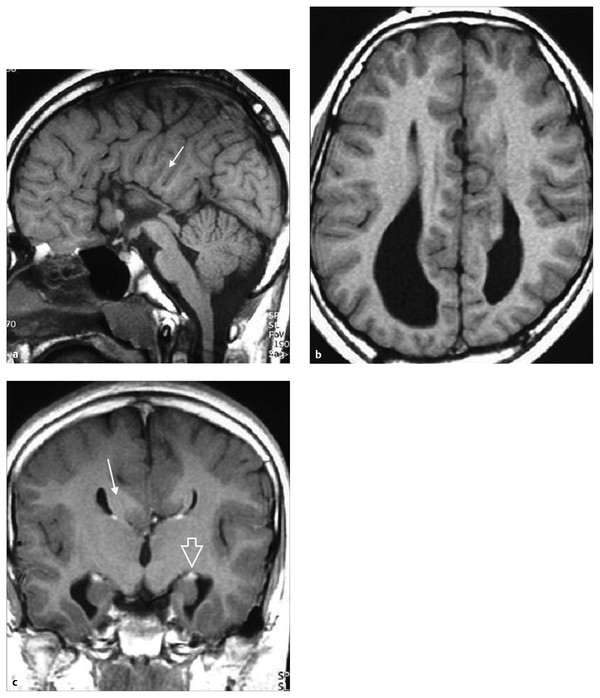
Fig. 15.6 Dysgenesis of the corpus callosum. (a) Midsagittal T1-weighted magnetic resonance (MR) image shows absence of the body, splenium, and rostrum of the corpus callosum. The interhemispheric gyri are oriented radially (arrow). (b) Axial T1-weighted MR image shows widely separated and parallel lateral ventricles. The frontal horns are pointed and the occipital horns dilated (colpocephaly). (c) Coronal T1-weighted MR image shows agenesis of the corpus callosum and Probst bundle (arrow) indenting the medial surface of the lateral ventricles, resembling a “moose head.” Associated malformations include vertical orientation of the hippocampus (open arrow) and distortion of the temporal horn.
ACC can sometimes be accompanied by interhemispheric cysts. Barkovich et al classified the cysts as type 1 if they communicate with the lateral or third ventricle and as type 2 if they do not.48 They are further divided depending on whether they are associated with hydrocephalus and/or other cerebral abnormalities, such as diencephalic malformation, heterotopia, and polymicrogyria. Except for the subgroup associated with Aicardi syndrome, ACC with interhemispheric cysts is seen predominantly in boys. The cysts are thought likely to be arachnoid, ependymal, or neuroepithelial, but their cause is still indeterminate.
Most children with isolated dysgenesis of the corpus callosum have only subtle neuropsychological deficits because of compensation by the anterior and hippocampal commissures, strengthening of the ipsilateral tracts, and possible bilateral representation of functions.41,44 Approximately 15% of patients with isolated ACC have cognitive deficits and psychological disorders, in addition to seizures and hydrocephalus. The morbidity of ACC is related to the associated intracranial anomalies and genetic syndromes.47,49
15.1.8 Microcephaly
Microcephaly is a clinical finding defined as a head circumference that is 2 or more standard deviations below the age- and sex-matched mean. Children with microcephaly do not have true dysmorphic facial features, but the small cranium creates the appearance of a sloping forehead and prominent ears. The small head reflects an undersized brain with a decreased number of neurons related to a decreased number of neural progenitors, decreased proliferation of neurons, or excess death of the progenitors and neurons.50,51 Environmental causes include congenital infections (e.g., rubella and cytomegalovirus infection); intrauterine exposure to teratogens (e.g., irradiation, alcohol, and cocaine); and intrauterine growth restriction. Microcephaly is also a feature of many other brain malformations, such as lissencephaly and HPE, and of metabolic disorders. When secondary causes of microcephaly and the presence other cerebral and extracerebral malformations are excluded, the children are then described as having either microcephaly vera (MV) or microcephaly with simplified gyral pattern (MSG). The two likely represent a continuous phenotype.
In MV, or “true” microcephaly, the small brain has a normal gyral pattern. The cortical mantle has the normal six layers, but it can be reduced in thickness, with a depletion of neurons in layers 2 and 3.50,51 MV has been attributed to faulty proliferation in the germinal zone before the 26th week of gestation, leading to exhaustion of the neuronal pool.52 Defects at multiple chromosomal loci, including the genes MCPH1, ASPM (MCPH5), and SLC25A19, have been identified and are transmitted in an autosomal-recessive manner.51 These children have intellectual disability that varies from mild to moderate. Seizures are uncommon, and the life span of patients with mild cases is often normal.
MSG carries a worse prognosis than MV. The child invariably has neonatal seizures, has moderate to severe intellectual disability, and often develops diffuse spasticity.53,54 Severe forms of MSG are lethal in infancy. Pathologic findings include a simplified gyral formation, but unlike in lissencephaly, the cortex is not thickened and has normal lamination. When thickening and dyslamination of the cortex are present, the diagnosis of microlissencephaly is given.55 MSG and microlissencephaly are possibly the result of a disruption of migration, as well as abnormal proliferation. Like MV, MSG is probably caused by defects of multiple genes transmitted in an autosomal-recessive manner.
15.1.9 Megalencephaly
Megalencephaly is defined as a brain volume greater than 2 standard deviations above the mean. Some authors suggest stricter criteria and include only patients with an occipitofrontal head circumference exceeding the mean by 2.5 standard deviations or the 98th percentile curve by 1 cm or more, but with normal ventricles and extracerebral space.56 Depending on the etiology, megalencephaly is described as anatomical or metabolic and is easily differentiated by the history and physical examination. Children with metabolic megalencephaly are born with a normal head circumference, which rapidly increases in girth during infancy from the brain cells’ accumulation of undegraded metabolic substrates or aberrant myelin due to a lysosomal deficiency or a dysmyelination syndrome.57 Consequently, the children develop progressive neurologic deterioration with signs and symptoms of elevated intracranial pressure (ICP). A thorough family history often reveals autosomal-recessive or X-linked–recessive inheritance.58 Examples of causes of metabolic megalencephaly are the gangliosidoses, mucopolysaccharidoses, sphingolipidoses, maple syrup urine disease, Alexander disease, and Canavan disease.
On the other hand, children with anatomical megalencephaly have normal ICP. The girth of the head, which may already be enlarged at birth, increases rapidly within first few months of life and then runs above, but parallel to, the 98th percentile curve.58 Almost all anatomical megalencephalies are inherited in an autosomal-dominant pattern. Some patients will have cutaneous stigmata and dysmorphic features consistent with certain neurocutaneous syndromes (e.g., neurofibromatosis and tuberous sclerosis) or rare growth disturbance disorders (e.g., achondroplasia).56 Cases without an etiology or associated syndrome are called primary or familial megalencephaly. These children have normal intelligence and neurologic examinations, and they often have relatives with large heads who are otherwise also normal.59 Although all the megalencephalies are classified by Barkovich et al as disorders of the proliferation of normal cells, there are no reported histologic studies of primary megalencephaly, and its embryogenesis is not known.60 Previous reports describing intellectual disability, epilepsy, paresis, and the presence of large cortical cells could be due to the grouping of all causes of megalencephaly together and confusion with hemimegalencephaly.56–58,61
15.1.10 Hemimegalencephaly
The clinical findings and prognoses of hemimegalencephaly are notably different from those of megalencephaly. There is marked asymmetry of the hemispheres in hemimegalencephaly, with the affected side enlarged and the opposite side slightly smaller than normal, resulting in varying degrees of midline shift (▶ Fig. 15.7). Occasionally, the ipsilateral cerebellum and brainstem are also increased in volume.62 Computed tomography (CT) and magnetic resonance (MR) imaging show ventriculomegaly with straightening of the frontal horns and colpocephaly in the affected hemisphere (▶ Fig. 15.8).63 The cerebrum is often lissencephalic, but polymicrogyria, gray matter heterotopia, focal cortical dysplasias, and rarely schizencephaly and ACC may also be seen. The cortex is thickened and is indistinct from the underlying white matter, which is often hyperintense on a T2-weighted MR image.
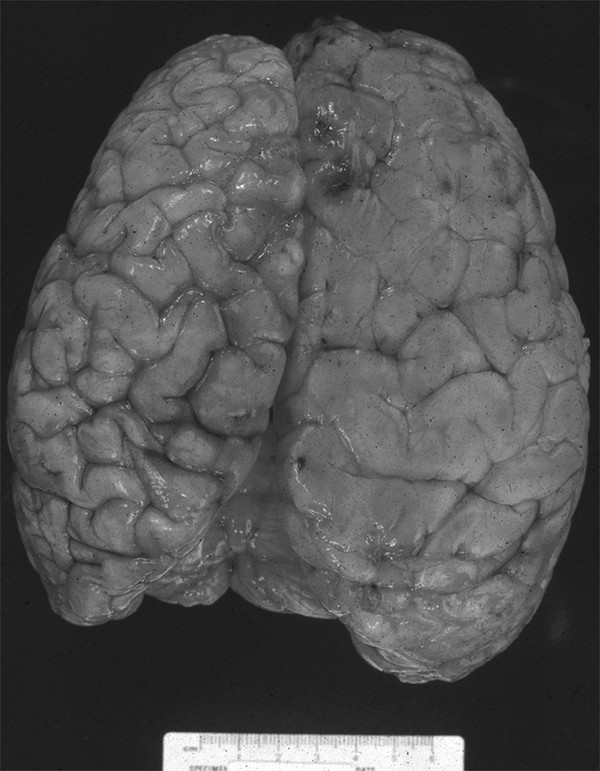
Fig. 15.7 Vertex view of the gross pathology of hemimegalencephaly. The entire right hemisphere is enlarged and has pachygyria and shallow sulci.
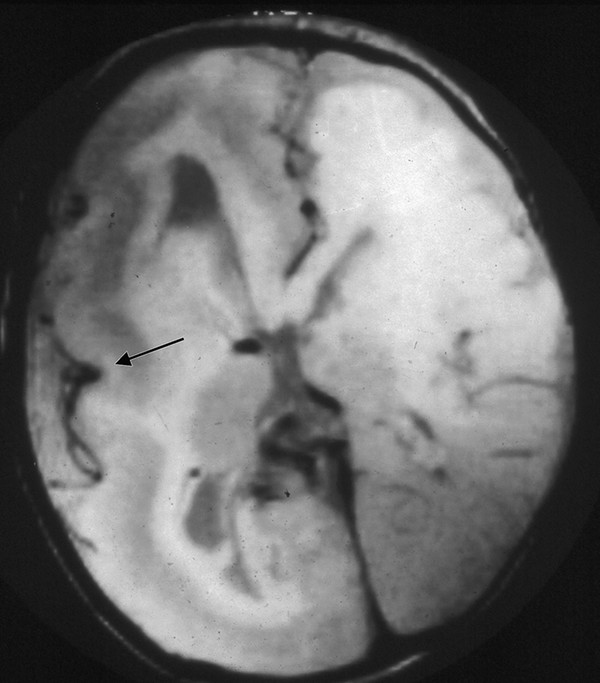
Fig. 15.8 Axial T1-weighted image of a child with hemimegalencephaly. The overgrown right hemisphere has an enlarged lateral ventricle, thickened cortex with a poor gray–white matter junction, and primitive veins in the sylvian fissure (arrow). There is diffuse loss of sulci in both hemispheres.
Histologic examination of the corresponding cerebrum reveals disruption of cortical lamination, a poor gray–white matter border, and abundant hypertrophic neurons and balloon cells positioned throughout the gray and white matter.64 Both cell types are immunoreactive to glial and/or neuronal proteins. Many authors have concluded that hemimegalencephaly is a disorder of cellular lineage, neuronal migration, and/or genetic regulation of body symmetry.63,65 However, the etiology is still uncertain, and there is no known genetic mutation or familial inheritance. Hemimegalencephaly with neurocutaneous disorders has a similar neurologic picture, pathologic features, MR imaging findings, and electroencephalographic abnormalities.63
The child with hemimegalencephaly typically presents with cranial asymmetry, hemiplegia, and intractable seizures that start soon after birth. Partial seizures occur most frequently, followed by tonic seizures and infantile spasms.63 The best chance for seizure control and improvement of neurologic and psychomotor development is with early hemispherectomy or hemispherotomy.64 Children with the early onset of neurologic symptoms demonstrate severe retardation of global development and an increased risk for mortality during infancy.63 At the other end of the spectrum, those with mild hemimegalencephaly may have only subtle deficits and a normal life expectancy.
15.1.11 Focal Cortical Dysplasia
Focal cortical dysplasias (FCDs) are localized areas of malformed cerebral cortex that are frequently associated with epilepsy. Taylor et al were the first to describe FCDs and their association with nonfamilial, nonsyndromic refractory epilepsy.66 A broad spectrum of histopathology is seen in the diagnosis of FCD. Currently, a three-tiered classification system is used to characterize specific clinicopathologic FCDs.67 Type 1 FCDs are isolated lesions that present as radial (FCD type 1a) or tangential (FCD type 1b) dyslamination of the neocortex, microscopically identified in one or multiple lobes. Type 2 FCD is an isolated lesion characterized by cortical dyslamination and dysmorphic neurons without (type 2a) or with (type 2b) balloon cells. Type 3 FCD occurs in combination with hippocampal sclerosis (FCD type 3a) or epilepsy-associated tumors (FCD type 3b), or adjacent to vascular malformations (type 3c) or epileptogenic lesions acquired in early life (type 3d), such as traumatic injury, ischemic injury, and encephalitis. The dysplasias can be localized to any part of the cerebrum but are frequently found at the frontal and temporal lobes.68
Type 2 FCD, with or without balloon cells, is usually visible on MR imaging, is highly epileptogenic, and has the lowest operative success rate. MR imaging findings include cortical thickening, poor delineation of the gray–white matter junction, and increased signal of the cortex and underlying white matter.69 Type 1 FCD is typically not visible on MR imaging but has better surgical outcomes than lesions with dysmorphic neurons.70 Besides the histologic composition, the seizure-free success rate is determined by the extent of resection.
The pathogenesis of FCD is not clear but may occur during cell proliferation, neuroblast migration and differentiation, and/or cortical organization.68 The tubers of tuberous sclerosis and cortices of hemimegalencephaly also contain balloon cells and are thus often grouped together with FCD.71
15.1.12 Congenital Neoplasms
Even though congenital neoplasms are classified under the heading of abnormal proliferation, their etiology is likely multifactorial, as is the case with postnatal-onset tumors. Particular to the developing brain, however, is its susceptibility to neoplastic transformation by genetic mutation and teratogens because of rapid cellular proliferation.72 Alterations of the regulatory cues for cell proliferation, differentiation, and apoptosis have the potential to result in neoplastic growth because many oncogenes and tumor suppressor genes involved in cancers are also regulators of normal CNS development.73,74 The finding of increased risk for pediatric brain tumors when there is a maternal family history of birth defects suggests either that malformations and neoplasia share a common etiology or that malformations confer a susceptibility for neoplastic transformation during childhood.75,76 Yachnis et al found that the granule cells that comprise vermian dysplasias are mitotically active after birth.77 These granule cells are therefore potential precursors of cerebellar tumors. Pediatric supratentorial tumors (e.g., ganglioglioma, dysembryoplastic neuroepithelial tumor, and pleomorphic xanthoastrocytoma) have also been linked to, or have been observed to develop from, cortical dysplasias.78–81
15.1.13 Lissencephaly
Lissencephaly is a diffuse bihemispheric abnormality that occurs in approximately 12 of every 1 million births and is characterized by a smooth or nearly smooth cortical surface (▶ Fig. 15.9).39,82 It is a disorder of neuronal migration, ranging in severity from the complete absence of convolutions (agyria) to a regional decrease in sulcation resulting in broad gyri (pachygyria). Lissencephaly is characterized by a cerebral cortex that is thickened to 10 to 20 mm (normal is 2.5 to 4 mm) and has four layers instead of six.83,84 Subcortical band heterotopia (SBH) is considered a mild form of lissencephaly in which bands of gray matter of varying width become embedded in the white matter and parallel the cortex of both hemispheres (▶ Fig. 15.10).
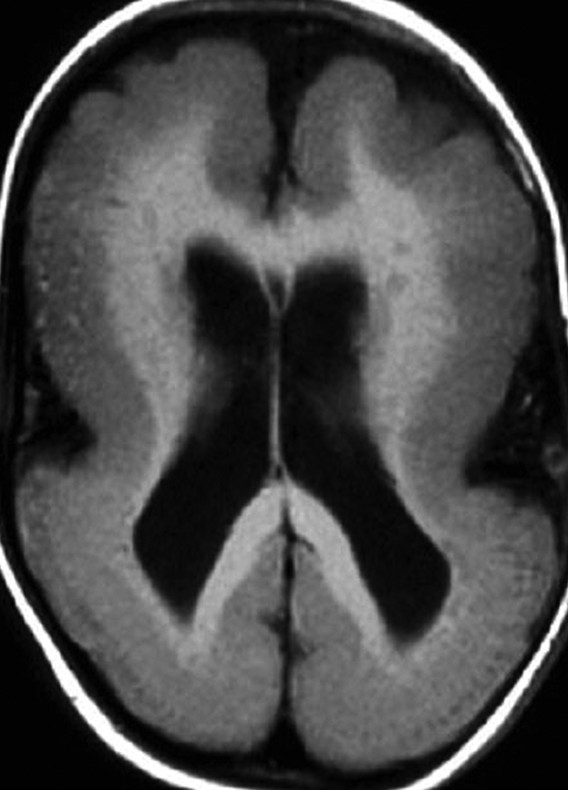
Fig. 15.9 Axial T1-weighted magnetic resonance image of classic lissencephaly (type 1) shows a smooth cortical surface with minimal gyri and sulci at the frontal lobes. The shallow sylvian fissures give the brain an “hourglass” configuration.
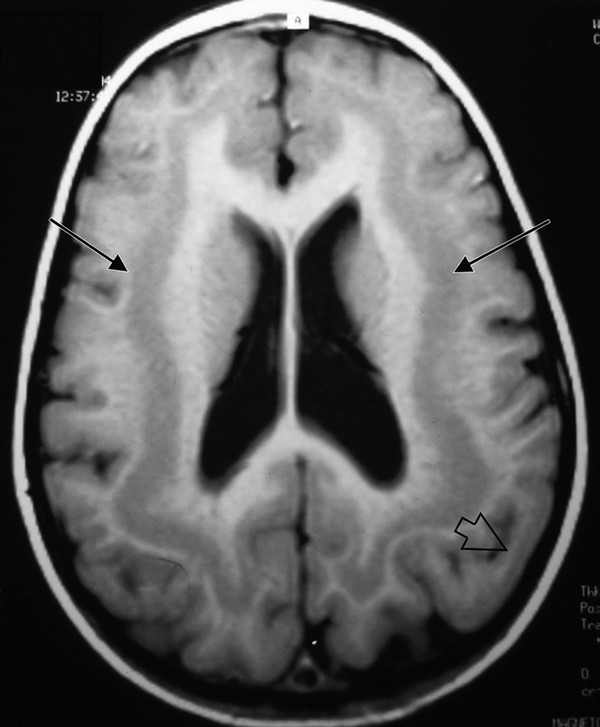
Fig. 15.10 Axial T1-weighted magnetic resonance image of a child with subcortical band heterotopia (double cortex). The bands of gray matter are situated between the lateral ventricles and cerebral cortex (arrows). Note the associated pachygyria (open arrow).
Lissencephaly can be classified genetically on the basis of genes involved in neuronal migration that, when mutated, result in lissencephaly. Classic lissencephaly (previously described as type 1) is associated with deletions of the LIS1 gene on chromosome 17p13 and the DCX (also known as XLIS) gene on chromosome Xq.85,86 Both genes regulate microtubule formation and activity during neuronal migration. The tubulin-α1a gene (TUBA1A), like LIS1 and DCX, encodes a microtubule-related protein and has recently been implicated in the pathogenesis of lissencephaly.86 Large deletions of LIS1 and the contiguous gene 14–3-3ε are associated with Miller-Dieker syndrome, which is characterized by lissencephaly with dysmorphic facial features.39,82 Patients with classic lissencephaly, on the other hand, have no other abnormalities, including those of the face. Mutations of DCX also cause isolated lissencephaly in males, but heterozygous females develop SBH.83 The frontal lobes of patients with DCX mutations are affected more than the occipital lobes, and the opposite is true in patients with LIS1 mutations.
Lissencephaly plus ACC occurs with mutation of the ARX gene located on chromosome Xp22.13 and is associated with underdevelopment of the genitalia.83 Patients who have lissencephaly with mutations of the RELN gene manifest malformations of the hippocampus and a severely hypoplastic cerebellum.83 Little is known about the genetics of microlissencephaly, which is characterized by a severely microcephalic brain with diffuse decreased sulci and cerebellar hypoplasia.54
Children with classic lissencephaly have a normal head size at birth, but their head growth plateaus, resulting in microcephaly within the first year.82 They are often intellectually disabled, have early-onset seizures, and exhibit hypotonia that shifts to hypertonia. The age at onset, severity of seizures, and degree of cognitive dysfunction depend upon the lobes of the brain involved and the volume and extent of misplaced gray matter.82 Treatment entails providing adequate nutrition, managing aspiration episodes, and controlling seizures. The majority of patients die within 10 years of aspiration pneumonia and sepsis.
15.1.14 Cobblestone Complex
Cobblestone dysplasia is a disorder of glycosylation that results in cortical dysplasia and dysmyelination, dysplastic cerebellum with cysts, and brainstem hypoplasia.87 Previously classified as lissencephaly type 2, because of the grossly smooth surface of the involved cortex, cobblestone dysplasia has now been re-categorized since its mechanism of malformation was elucidated. In contrast to lissencephaly, in which the neurons fail to reach the cortical plate, in cobblestone dysplasia, the neurons pass through the cortex into the subpial space and appear on the brain surface like cobblestones.83,87 The cortex is thick, as it is in lissencephaly, but cortical lamination is not evident. Moore et al discovered that failure to glycosylate the dystroglycan results in breaches in the glia limitans, the pial surface lamina, allowing migrating neurons to exit the brain proper.88
Loss of dystroglycan function may underlie the cobblestone dysplasia observed in at least three genetic disorders: Walker-Warburg syndrome, Fukuyama-type congenital muscular dystrophy, and muscle–eye–brain disease.89 Children born with these congenital muscular dystrophies have profound neonatal hypotonia, ophthalmologic malformations, and cerebellar dysplasia.83 Patients may die at infancy or live to adulthood without significant morbidity depending on the severity of weakness, seizures, and intellectual disability and on the type of muscular dystrophy.
15.1.15 Gray Matter Heterotopia
Neuronal heterotopias are disorders of neuronal migration that result in collections of normal neurons situated anywhere between the germinal zones and the subcortical white matter.90 Normal neural migration entails attachment of neurons to radial glial cells and migration along the glial cells, followed by detachment at the appropriate cortical layer. Interruption of this basic process along various points results in the heterogeneous appearances of the neuronal heterotopias.83 Barkovich et al grouped heterotopias into three categories: periventricular (subependymal), subcortical (other than band heterotopia), and marginal glioneuronal heterotopia.60
Periventricular heterotopia is characterized by nodular masses of gray matter lining the lateral ventricles and protruding into the ventricular spaces (▶ Fig. 15.11). Bilateral periventricular nodular heterotopia may be X-linked, and therefore familial, while unilateral subependymal heterotopia and sparsely scattered subependymal heterotopia present sporadically.91 When it is X-linked, bilateral periventricular nodular heterotopia is more prevalent in females because of its lethality in males. Bilateral periventricular nodular heterotopia results from a defect in the filamin A (also known as filamin-1) gene (FLNA), which is needed for linking actin filaments to glycoproteins and is essential to the process of neuronal migration.92,93 Ninety percent of these patients have epilepsy, and many have intractable seizures.39 Females who are heterozygous for the mutation have epilepsy, which varies in severity, and have borderline to normal intelligence. Patients who have periventricular heterotopias are more likely to demonstrate normal development in comparison with those who have other types of heterotopia.
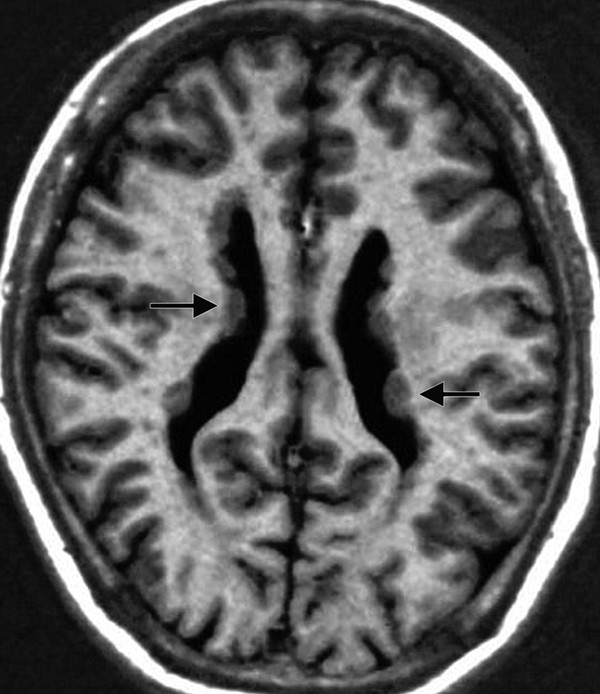
Fig. 15.11 Axial T1-weighted magnetic resonance image of bilateral periventricular nodular heterotopia shows gray matter lining and indenting the lateral ventricles (arrows).









