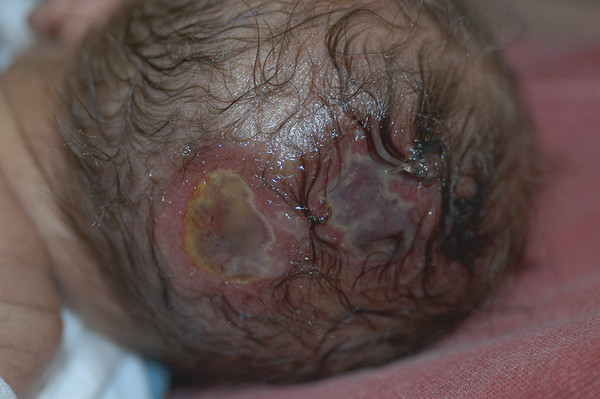
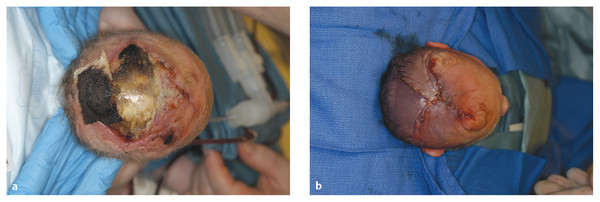
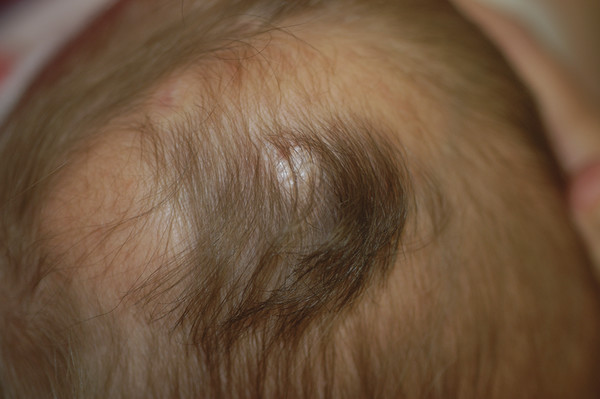
Congenital Lesions of the Scalp and Skull Congenital lesions of the skull and scalp are common, and the vast majority of them are benign. Nevertheless, the treatment of these lesions requires a good understanding of their natural history, radiographic findings, diagnostic criteria, and treatment options. Treatment ranges from observation to multidisciplinary care involving multiple specialists. Although we cannot cover all the lesions, we attempt to discuss some of them that are seen commonly in a pediatric neurosurgical practice. In particular, we discuss three commonly encountered entities: aplasia cutis congenita, atretic cephaloceles, and parietal foramina/cranium bifidum. Aplasia cutis congenita (ACC), also referred to as cutis aplasia, is a heterogeneous condition in which one or more components of the scalp and/or skull are absent at birth. Histologically, cutis aplasia is characterized by a lack of epidermis, dermis, fat, and in severe cases muscle and/or bone (▶ Fig. 18.1). Although the condition was first described in the extremities by Cordon in 1767,1 and any part of the body can be affected, 85% of cases involve the scalp.2–7 ACC of the scalp was first described in 1826 by Campbell,8 and Billard later reported a concurrent cranial defect.3,8–10 Since that time, there have been hundreds of reports of this condition.3 Fig. 18.1 Aplasia cutis congenita in a newborn with full-thickness defect and exposed dura. ACC is reported to occur in about 1 in every 10,000 live births and is more common in females than in males.2,11 Nevertheless, ACC is likely underreported because mild cases may be confused with other scalp conditions.2 The cranium and dura are affected in about 15% to 30% of cases2 (references 5, 6, 8, and 9 in Bharti et al2), and these cases require acute treatment. Dessication of the dura, especially over the superior sagittal sinus, can result in significant morbidity or death. A number of theories have been posited over the years to explain the cause of this condition. The etiology seems to be multifactorial, as no single theory seems to explain ACC fully. As with many conditions, both genetic and environmental factors appear to be related to ACC. Most cases of ACC occur sporadically, but there is a family history in 40% of affected children. The majority of hereditary cases of ACC are passed in an autosomal-dominant fashion,12–16 although autosomal-recessive inheritance has also been reported13,14,17,18 Multiple syndromes show association with ACC. One of the best known is Adams-Oliver syndrome. Children with Adams-Oliver syndrome can have ACC, with or without involvement of the skull or dura. In addition, these children may have shortened or absent fingers or toes, loss of the metacarpals, absence of the lower extremities below the calf, and mottling of the skin. Adams-Oliver syndrome is thought to be inherited in an autosomal-dominant pattern.2,13,19,20 Other syndromes associated with ACC include Setleis syndrome, Anderson-Hollister-Szalay syndrome, Johanson-Blizzard syndrome, and Goltz syndrome.2,5,21,22 A number of environmental factors have also been cited as possible causes of ACC. According to one theory, amniotic adherences to the skin of the fetus tear off, leaving areas devoid of skin. This theory seemed to have been disproven, at least in most cases, because most examined cases showed normal placentas.3,23 Teratogens have also been implicated in causing ACC. These include heroin, alcohol, cocaine, methotrexate, antithyroid medications, angiotensin-converting enzyme (ACE) inhibitors, and other medications.2,5,6,24 Another possible mechanism is tension on the fetal skin during development. It is thought that tension on the vertex, which sustains the greatest amount of stretch during head growth, can disrupt the skin, leading to ACC.2,5,24,25 Other factors that have been proposed as possible mechanisms leading to ACC include vascular disruption of the fetus leading to ischemic injury2,4,6,22,24,25, intrauterine infection, and intrauterine trauma.2,6,26 Of note, Ingalls in 1933 found that there was an associated fetus papyraceus, or mummified dead fetus, at the delivery of many newborns with ACC of the trunk or limbs.3,27 As one can see from this myriad of possible causes, there is no unifying theory to explain ACC. Rather, it is thought to be multifactorial in etiology, with both genetic and environmental causes. Different classification schemes have been proposed to describe the disease. Frieden devised a nine-part classification system based on area of the body affected, other abnormalities, and inheritance pattern.3 Lesions of ACC do not have the normal histology of stratified squamous epithelium of the scalp. Instead, they show a loose fibrous stroma devoid of hair follicles, epithelium, and sweat glands. They are covered by a thin epithelial membrane with a thickness of one cell layer.13,28–30 The presentation of ACC can vary greatly, and the management of a lesion requires a careful examination and understanding of its extent. More than 80% of lesions are located at or near the midline, and many are situated close to the parietal hair whorl.3,13,28,31 These lesions can present in all sizes. Most are less than 2.5 cm in diameter, but some can be more than 8 cm in diameter.13,32 Larger lesions, and lesions that involve the skull and dura, can pose significant challenges to treatment. Many cases of ACC present as a hairless patch of thin skin over the vertex. Small hairless patches can sometimes be confused with conditions other than ACC. ACC affects the skull or dura in about 15 to 30% of cases2 (references 5, 6, 8, and 9 in Bharti et al2). Serious morbidity can result if the dura of the superior sagittal sinus is exposed. The exposure of underlying structures can lead to infection, sagittal sinus thrombosis, and/or major hemorrhage from the superior sagittal sinus, which can even lead to death. Hemorrhage can occur if the sinus becomes dry and brittle.2 Indeed, one of the cases first described by Campbell was that of an infant who died after bleeding from an exposed superior sagittal sinus.8 This rare but known scenario must be recognized by the physician in order to guide treatment and prevent major morbidity (▶ Fig. 18.2). Fig. 18.2 (a) Aplasia cutis congenita with desiccated dura and exposed superior sagittal sinus. This child had a hemorrhage from the superior sagittal sinus shortly after presentation in the intensive care unit and required emergency rotation flap closure. (b) Photograph after closure of defect. The treatment of ACC is based on the size and location of the lesions.2 Fortunately, most defects are small and relatively superficial. These are effectively managed with local wound care. Large areas with exposed dura can also be safely managed without surgery, but the potential morbidity of these lesions is much greater, and treatment must be monitored carefully.6 Conservative treatment requires maintaining a moist environment at all times. Typical dressings include hydrocolloid, petrolatum with or without antibiotic ointment, bacitracin, and silver sulfadiazine. Many authors advocate topical antibiotic ointments to provide both a moist wound-healing environment and, conceptually, prophylaxis against infection and meningitis. If a topical ointment is applied, it should be applied in a thin coat to reduce buildup and reapplied only as needed to maintain a moist wound bed. It is important to clean gently any caked or dry emollient from the wound because this layer can create a barrier to the beneficial effects of subsequent medication. Adjuvant treatment with oral or intravenous antibiotics is controversial. There have been reports of adding growth factors (e.g., recombinant human fibroblast growth factor) to the wound to promote further healing, although the need for this is dubious.33 If larger areas of dura are exposed (> 2 cm2) or circumstances prevent reliable outpatient compliance with dressing (e.g., dysfunctional family dynamics), we strongly recommend hospital admission until the dura is completely covered with granulation tissue. In a neonate, this occurs very rapidly and reduces the likelihood of preventable complications. Some authors advocate early treatment with allograft and autograft to reduce the scarring associated with spontaneous healing. Apligraf (Novartis, Basel, Switzerland) is a bilayered skin construct formed from allogeneic foreskin keratinocytes and a dermal layer consisting of bovine collagen lattice and human fibroblasts. It has been used but patchy contractures still result, likely from reaction to the bovine component.34 Other skin substitutes, such as those made by Integra (Plainsboro, NJ), have been reported to yield good results.35 The placement of cultured epithelial autografts on the surface of AlloDerm (KCI Lifecell, San Antonio, TX) has also been tried, although the aesthetic outcomes are unknown.36 Coverage with autograft or allograft material may achieve earlier closure of the wounds, but it is unclear if and how these methods can reduce scarring. In our opinion, the lack of evidence to support the superiority of these treatment options over more conservative measures does not justify their relatively high cost. Although even the largest areas of deficiency will heal if a clean, moist wound environment is maintained, conservative treatment requires constant surveillance and a well-trained and motivated ancillary staff. If this level of care is not available and prolonged observation with local wound care is impractical, one may consider early wound closure to ensure a safer and more predictable outcome, especially if the dura overlying the sagittal sinus is exposed, which can lead to hemorrhage and death. Early closure can be achieved by using various skin substitutes, as noted above, or by rotating local tissue flaps.25,37 The benefit of early closure must be weighed against the potential pitfalls. For example, grafted material can fail to adhere or become infected. Moreover, grafts may be difficult to separate from the underlying dura if later scar excision is planned. Rotational flaps in infants can undergo partial or complete necrosis. Surgical delay should be considered if possible before larger, complex, or multiple-pedicle reconstructions. Some authors have demonstrated good results with the use of composite bone and soft-tissue flaps at a very early age.38 Defects that extend into the deep dermis will leave a scar, and hair will not regrow in these areas. In such instances, many families express an interest in reducing or removing the scar. The timing of this depends on the size of the area of alopecia and whether any underlying bone defect is present. If it is, we recommend deferring any soft-tissue reconstructive procedure until the bone has healed or until the child is at least 2 years of age because little osseous healing can be expected after this time. The best method of soft-tissue and/or bone reconstruction depends on the size of the defects. Areas of alopecia with a width of up to 2 cm can be addressed with simple excision and closure. Larger areas up to 4 cm in width can also be closed with this method, but more sophisticated techniques are required to mobilize the scalp. Specifically, the scalp must be widely undermined and the galea aggressively scored with multiple releasing incisions. When the ability to close the defect fully is in question, it is advisable to begin with only a single incision along one edge. After mobilization, the tissue excision can be tested by overlapping the edges of the wound and excising the redundant tissue. If the entire scar cannot be excised, the wound is closed, and one can come back in 6 months to a year to complete the excision. This method of serial excision is very effective and can address even very large areas of alopecia. In rare instances in which the area of alopecia is extensive, tissue expansion or rotational flaps should be considered.9 The former option requires two procedures and has potential complications, such as infection and expander extrusion or rupture. In very young infants, it also can deform the cranium. The latter option works well if carefully planned. The use of surgical delay is advisable in most instances. Delay involves performing all incisions and elevating, but not rotating or in-setting, the flaps several weeks before the definitive reconstruction. This step significantly improves the perfusion and durability of soft-tissue flaps, especially those with a high length-to-width ratio. It is advisable to seek the help of an experienced reconstructive surgeon before embarking on this treatment path. Bone defects that are large enough to pose a safety concern (>2 cm2) may warrant grafting. In most cases, the overlying soft tissue is of poor quality, and bony grafting should be done concurrently with the soft-tissue reconstruction to ensure a healthy, vascularized covering for the graft. This can be done with corticocancellous particulate autograft in a younger child or split cranial bone if the child is older.39 We advise against the use of alloplastic materials in pediatric cranial reconstruction because they have higher rates of extrusion and infection than autografts, are very expensive, rarely osseointegrate, and do not grow with the cranium. There are few if any indications for alloplastic cranial reconstruction in the pediatric population. Cephaloceles are generally defined as herniations of structures of the cranium through a defect in the skull, and they can have a fairly diverse presentation with associated abnormalities of the brain.40,41 Three major types of cephaloceles include encephaloceles, meningoceles, and atretic cephaloceles. These lesions are often defined by their location as either occipital or parietal.42 Parietal cephaloceles have been described as having a worse prognosis,42 but this has been contradicted elsewhere.41 Encephaloceles (or meningoencephaloceles) are herniations of the brain and meninges, and children with these malformations often have the worst prognosis for cognitive development. Meningoceles are herniations of meninges that do not contain cerebral tissue. These lesions are filled with cerebrospinal fluid (CSF) that communicates with the underlying brain via a connection. Atretic cephaloceles have been described differently by various sources, which can be quite confusing. In general, an atretic cephalocele is a congenital lesion of the scalp and skull consisting of a skin-covered dural sac that may or may not contain fluid. Each lesion generally has a stalk that connects it to the underlying dura. Atretic cephaloceles can be either flat or cystic. We distinguish cystic atretic cephaloceles from meningoceles in that the stalk of an atretic cephalocele usually has a closed lumen, and the cyst fluid does not freely communicate with the underlying brain. On the other hand, meningoceles contain CSF that communicates with the underlying brain. These lesions can contain rests of glial tissue in addition to meninges.41,43 Some may think of the lesions as “burnt-out” or sequestered cephaloceles.44 Because they contain neural tissue, they have also been referred to as heterotopic neural nodules.44 Other authors have used the terms rudimentary cephaloceles, occult cephaloceles, and abortive cephaloceles.41–43,45,46 Atretic cephaloceles have been reported in various studies to comprise 2542 to 50%41 of all cephaloceles. Most atretic cephaloceles are thought to be sporadic, but some have be seen in association with underlying genetic syndromes. Walker-Warburg syndrome, or cerebro-ocular dysgenesis–muscular dystrophy syndrome, is a rare autosomal-recessive form of muscular dystrophy.47 It is associated with multiple cerebral anomalies, including cephaloceles. A number of other genetic syndromes or familial associations have been described with atretic cephaloceles.48–51 In addition, some teratogens have been linked to cephalocele formation.41 Multiple theories have been proposed to explain the formation of atretic cephaloceles. Some believe that they form when the neural tube fails to close. Others believe that the neural tube closes, then reopens and develops abnormally.41 Drapkin proposed that atretic cephaloceles form from remnants of the neural crest after faulty migration.43 Yokota et al believed that an atretic cephalocele forms after the involution of a true meningocele in utero.42 Atretic cephaloceles generally present at or near the midline in the parietal or occipital regions, often in the vicinity of the posterior fontanel.43 These lesions can present as a flat bump or as a cystic structure (▶ Fig. 18.3 and ▶ Fig. 18.4). They are usually skin-covered, but the skin can be quite thin and may be ulcerated or discolored with a vascular stain.41,44 The lesions are often hairless, but a thick ring of hair may surround the lesion, often called the “hair collar” sign.52 There is generally an associated bone defect. In one study, the lesions ranged in size from 10 to 35 mm.41 Fig. 18.3 Atretic cephalocele with the “hair collar” sign and capillary stain.
18.1 Aplasia Cutis Congenita
18.1.1 Background
18.1.2 Causes
18.1.3 Pathology
18.1.4 Clinical Presentation
18.1.5 Treatment
18.2 Atretic Cephaloceles
18.2.1 Background
18.2.2 Causes
18.2.3 Clinical Presentation
Congenital Lesions of the Scalp and Skull
Only gold members can continue reading. Log In or Register to continue







Full access? Get Clinical Tree


