Congenital Vertebral Anomalies
Congenital vertebral anomalies are relatively common disorders, ranging from simple, asymptomatic “block” vertebrae to complex combinations of anomalies involving multiple vertebral levels. They can occur anywhere from the craniovertebral junction to the coccyx. This chapter covers congential vertebral anomalies from the cervical spine to the sacrum. Congenital vertebral anomalies may be associated with other birth defects, and awareness of these associations is important. To manage patients with congenital vertebral anomalies effectively, a firm grasp of the relevant embryology, classification, natural history, clinical evaluation, and treatment principles is essential.
29.1 Historical Background
Although spinal deformities have been recognized in the medical literature for centuries, it was not until the early part of the 20th century that the first vertebral anomaly syndromes were characterized. In 1912, Klippel and Feil reported a patient with the triad of low posterior hairline, short neck, and reduction in cervical spine motion, thus providing the first description of the syndrome that now bears their names.1 A little over a decade later, Foix and Hillemand provided a classification scheme for caudal agenesis based on the extent of sacral and coccygeal involvement.2 Significant advancements in the description and understanding of vertebral anomalies occurred with the development of spinal radiographs in the middle of the century, and extensive effort over the past 20 years by many authors, notably Winter et al,3,4 McMaster and Ohtsuka,5 and Pang et al,6–9 has provided tremendous insight into the pathogenesis, treatment, and prognosis of these disorders.
29.2 Embryogenesis
The sequential stages of the development of the spine have been extensively discussed in prior texts7 and will be only briefly reviewed here. The first six embryonic weeks set the stage for early vertebral development in a process known as primary neurulation. In that period, also known as the mesenchymal stage, the notochord develops in the first few embryonic weeks from cells within the Hensen node.10 Somitic mesodermal cells migrate from a position just caudal to the Hensen node and ultimately lie just lateral to the midline notochord. These cells coalesce into paired somites in a rostral to caudal direction, all under the influence of the notochord, the neural tube, homeobox genes, and cell adhesion proteins.11–13 The somites ultimately divide into sclerotomes and dermomyotomes, with each of the bilaterally paired sclerotomes giving rise to a single vertebral body and single set of posterior elements during a migrational phase in the fourth and fifth embryonic weeks.12–14
Although this process of primary neurulation accounts for the formation of the vertebral column and spinal cord down to the lumbosacral junction, most sacral and coccygeal vertebrae develop from the caudal eminence in a poorly understood process called secondary neurulation.10 The caudal eminence is a mass of undifferentiated cells at the caudal end of the primary neural tube.6 Secondary neurulation begins at approximately the fourth embryonic week and is responsible for the formation of the spinal cord, nerve roots, and vertebral column of the sacral and coccygeal areas.15,16 New insights into the complex manifestations of secondary neurulation are provided by Pang et al.8,9
During the sixth embryonic week, the chondrification stage begins with the formation of three paired chondrification centers within a vertebra. One set of chondrification centers develops anteriorly to form the vertebral body, and two sets develop posteriorly to form the posterior elements. This process develops in a cranial and caudal direction from the cervicothoracic junction and is responsible for rapid vertebral column growth.17 It culminates at approximately the ninth embryonic week. Ossification of the cartilaginous elements begins shortly thereafter and ultimately ends somewhere between the 14th and 18th years of life.18,19 The intervertebral disk develops from tissue derived from perinotochordal mesenchymal cells and begins its formation in the chondrification stage.12
29.3 Epidemiology
Accurate statistics regarding congenital vertebral anomalies are difficult to come by because many patients are asymptomatic and the anomalies remain undiscovered. Incidence rates are also difficult to gather because many cases are discovered incidentally and never evaluated further. Furthermore, estimates are difficult to obtain in young children because of radiographic variations. At an early age, vertebrae have partially ossified centra and arches that may be confused with congenital vertebral anomalies.18,20
An estimated 5% of fetuses have vertebral anomalies.21 About 3% of the normal population have one or two more than the usual 7 cervical, 12 thoracic, 5 lumbar, and 5 sacral vertebrae; approximately 2% of the population have one less.18 About 7 in 1,000 individuals in the general population have a congenital union of two or more cervical vertebrae.22,23 The incidence of caudal agenesis in Sweden was reported as 1 in 7,500 births, a number similar in magnitude to the 1 in 4,000 incidence found in Chicago.24 The incidence of scoliosis of more than 10 degrees was reported as a little over 1% for students in Beijing,25 but a slightly higher figure of 2 to 3% has been cited for idiopathic scoliosis in skeletally immature children in the United States.26
29.4 Etiology
Congenital vertebral anomalies commonly result from abnormal development during the first trimester of pregnancy.27 The nature and timing of the insult to the embryonic vertebral column determine the type of congenital abnormality that is produced. Hemivertebrae and hypoplastic vertebrae arise during the mesenchymal stage, with a disruption of the primary chondrification centers and a pairing defect of the responsible sclerotomes, respectively.17 Some authors have proposed that vertebral body hypoplasia and aplasia, both common causes of kyphotic abnormalities, arise during the chondrification stage, possibly as a consequence of absent centrum vascularization.17,19,28 Environmental factors, including infections such as tuberculosis, can also result in hypoplastic vertebrae and congenital scoliosis.27
The etiology of osseous malformations in the cervical spine is probably multifactorial.29 Autosomal-dominant inheritance of cervical ribs has been reported,30 and the congenital cervical fusions occurring with abnormalities such as the Apert and Crouzon syndromes are probably based on autosomal-recessive and autosomal-dominant inheritance patterns, respectively.31 Genetic inheritance does not account for many of these lesions, however, and there is some evidence to suggest that congenital vertebral anomalies result from vascular occlusions during development.
Some authors have suggested a subclavian artery supply disruption sequence to explain the pathogenesis of Klippel-Feil and other vertebral anomalies.29,32 They hypothesize that cervical fusions result from the disruption of intersegmental vessels arising from the vertebral arteries at the time of resegmentation of the sclerotomes. Specific refinements of this hypothesis have been forwarded by Tredwell et al,33 who reported that the fetal alcohol syndrome variant of the Klippel-Feil anomaly is always associated with a single level of congenital fusion. In contrast, other cases of Klippel-Feil have only a 20% rate of single-level fusion. They relate this finding to a specific teratogenic insult occurring between the 24th and 28th days of embryonic life.33
Vascular occlusion may be only one of several causes of congenital vertebral anomalies. Chandraraj34 described anatomical specimens in which failure of development of the zygapophyseal joint appeared to cause vertebral body fusions, or block vertebrae. He suggested that in some cases, the condition was probably linked to a defect of an inductor substance (e.g., the neuraxis) that influences normal morphogenesis of the vertebral arch in the embryonic period.
The etiology of more complex vertebral anomalies, such as congenital vertebral dislocation, segmental spinal dysgenesis, and medial spinal aplasia, is currently poorly understood.3,16,35,36 Dias and colleagues35 have proposed that congenital vertebral dislocation arises as a result of early embryonic buckling that affects all vertebral elements in a segmental fashion between the fourth and sixth embryonic weeks.
29.5 Classification
Congenital anomalies of the spine have previously been classified according to their anatomy, pathology, or embryology.20,37,38 Because a comprehensive classification scheme encompassing all types of vertebral anomalies is not available, they have traditionally been divided into disorders of vertebral formation, vertebral segmentation, or a combination of both (see box ▶ “Classification of Congenital Vertebral Anomalies”). Many of the underlying genetic and embryologic abnormalities are only beginning to be understood. Our understanding of many disorders, including congenital vertebral dislocation, segmental spinal dysgenesis, and medial spinal aplasia, is evolving.16,35,36,39 This chapter relies primarily on the traditional classification scheme and discusses the findings of the more complex disorders in a separate section.
Classification of Congenital Vertebral Anomalies
Disorders of formation
Wedge vertebrae
Hemivertebrae
Caudal agenesis
OEIS syndrome
VACTERL syndrome
Disorders of segmentation
Block vertebrae
Segmental bars
Combination disorders
Special disorders
Congenital vertebral dislocation (deformation disorder)
Segmental spinal dysgenesis (probable disorder of formation)
Medial spinal aplasia (probable disorder of formation)
Abbreviations: OEIS, omphalocele, cloacal exstrophy, imperforate anus, and spinal deformities; VACTERL, vertebral anomalies, anorectal malformations, cardiac malformations, tracheoesophageal fistula, renal anomalies, and limb anomalies.
Disorders of vertebral formation are regarded as failures in development of any part of the vertebral column. They may be either complete or partial and may be unilateral or bilateral. Incomplete formation of a vertebral body results in a wedge vertebra, with one side hypoplastic and with an asymmetric appearance. If one pedicle and the adjacent vertebral body are absent, a hemivertebra results. A hemivertebra can be further classified depending on whether it is fused to one or both adjacent vertebrae. An unsegmented hemivertebra is fused to the adjacent vertebrae above and below, a partially segmented hemivertebra is fused to one vertebra above or below, and a segmented hemivertebra is separated by a disk space from each adjacent vertebra. In the sacrococcygeal area, a more complex example of these disorders is the caudal agenesis syndrome.
Disorders of vertebral segmentation give rise to failures of vertebral separation and different degrees of intersegmental fusion (▶ Fig. 29.1). These disorders include block vertebrae and unilateral unsegmented bars. Block vertebrae may be the best example of a segmentation failure resulting from the failure of a somite segment to separate into cephalic and caudal halves, creating one large block with no intervening disk. A similar defect occurring on one side of the developing spinal column results in the formation of a unilateral unsegmented bar. The term unsegmented bar describes a bony bar that fuses the disk space and facet joints of one or more adjacent vertebral levels. The fusion may exist in the anterior spinal column, posterior spinal column, or both and may exist alone or in combination with other disorders. Vertebral growth proceeds on the segmented side only, a condition that often leads to severe scoliosis. A segmentation failure across the anterior portion of adjacent segments with normal development of the posterior portion of the vertebra leads to progressive kyphosis. Lateral segmentation anomalies are presumed to begin in the membranous and cartilaginous phases of vertebral development.17 In contrast, vertebral body segmentation anomalies are thought to arise as a result of disordered ossification.17,35,39
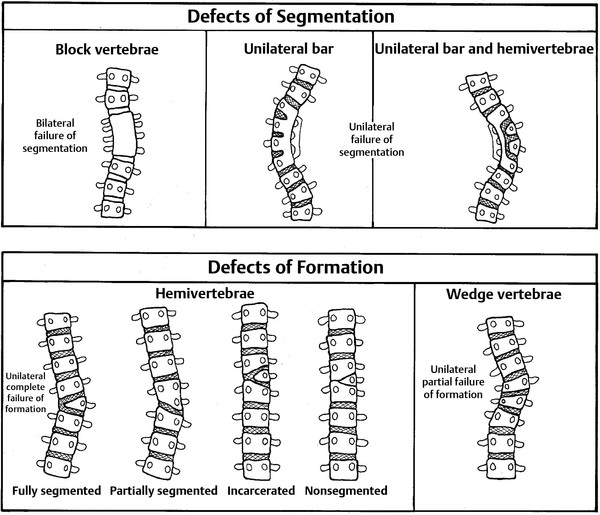
Fig. 29.1 Schematic drawings representing defects of segmentation and formation.
Recent work has described a smaller, significant group of complex congenital vertebral anomalies that result in either craniocervical deformity and instability or congenital kyphoses (see box ▶ “Special Congenital Vertebral Anomalies”).35,36,39 These disorders include atlantoaxial hemivertebrae, congenital vertebral dislocation, segmental spinal dysgenesis, and medial spinal aplasia. Because it is unclear whether they can be explained by the traditional classification system, they are placed together under a separate heading. An atlantal hemiring is probably the most common cervical congenital vertebral anomaly seen by pediatric neurosurgeons.40 Atlantal hemiring is defined as a bony discontinuity of the C1 ring in conjunction with the lateral displacement of the C1 lateral masses. It is frequently associated with occipital condyle abnormalities and absence of the transverse ligament, and it often leads to significant craniovertebral instability
Special Congenital Vertebral Anomalies
Congenital vertebral dislocation
Involved vertebrae malformed
Superior vertebrae: elongated posterior elements with abnormally large spinal canal, bifid or incomplete laminae
Inferior vertebrae: misshapen, may be either smaller or larger than normal, posterior elements typically normal, spinal canal normal at this level
Spinal cord: intact, displaced by bony elements
Atlantal hemiring
Bony discontinuity of the C1 ring in conjunction with lateral displacement of the C1 lateral masses
Associated with occipital condyle abnormalities and absence of the transverse ligament
Often causes or leads to significant craniovertebral instability
Segmental spinal dysgenesis
Multilevel congenital spinal stenosis, hourglass shape of spinal canal
Absent pedicles, neurocentral junctions and transverse processes at involved levels
Stenotic posterior osseous ring encircling the spinal cord, separated from the posterior vertebral cortex by fat-filled space
Absent nerve roots at level of the stenosis
Normal vertebrae above and below the malformation
Spinal cord present cranial and caudal to the malformation
Generally normal sensorimotor function or incomplete neurologic deficits
High incidence of associated anomalies: tethered spinal cord, equinovarus deformities, Klippel-Feil syndrome, crossover rib, renal agenesis or duplication, situs inversus, and tetralogy of Fallot
Medial spinal aplasia
Segmental or suspended agenesis of between 3 and 11 thoracic and/or lumbar vertebrae without associated lumbosacral agenesis
Spinal cord agenesis caudal to the malformation
Complete congenital paraplegia below the level of malformation
Severe orthopedic deformities with “Buddha-like” posture
29.6 Natural History
Because of their relatively high incidence, a good deal of information is known about the natural history of scoliotic and kyphotic curves in patients with congenital vertebral anomalies. This information, coupled with knowledge of normal and abnormal spinal growth, can be used to make decisions about the need for and timing of surgical intervention.
The growth potential associated with hemivertebrae is determined by whether the defect is segmented, partially segmented, nonsegmented, or incarcerated.5 A fully segmented hemivertebra has normal disk spaces above and below the affected level and therefore has the potential for unopposed longitudinal growth. Isolated segmented hemivertebrae in the thoracic spine produce progressive curves with a deterioration rate of 1 to 2 degrees per year. If multiple hemivertebrae exist, the rate of deterioration can be as high as 5 degrees per year. Lumbosacral hemivertebrae produce oblique spinal deformities, resulting in marked pelvic obliqueness and significant compensatory curves in the thoracolumbar spine. A partially segmented hemivertebra is fused with one adjacent vertebra and has less potential to produce significant deformity. A nonsegmented hemivertebra is fused to the vertebrae above and below and therefore has no potential for growth. In these cases, progressive deformity does not occur, and treatment is rarely required. Incarcerated hemivertebrae are ovoid and have narrow growth plates with little growth potential. These hemivertebrae appear to be contained by the vertebrae above and below and are rarely associated with significant deformity.
Unilateral unsegmented bars produce a unilateral growth tether and scoliosis. Isolated unilateral unsegmented bars in the thoracic spine have the potential to deteriorate at a rate of 5 degrees per year and require treatment. If bilateral failure of segmentation occurs, producing block vertebrae, then the disk spaces are symmetrically fused and therefore have little potential for producing spine deformities.
Combination deformities involving failures of both vertebral formation and segmentation are often very complex and have an increased potential for causing significant spinal deformity. The combination of a unilateral unsegmented bar with one or more contralateral hemivertebrae, for example, can lead to a rapidly deteriorating thoracolumbar scoliosis that has the potential to progress at 5 to 10 degrees per year. In many cases, these combination anomalies are fused at the time of diagnosis to prevent severe, intractable deformities in the future.
More is known about the natural history of thoracolumbar scoliosis than any other congenital vertebral anomaly. According to McMaster and Ohtsuka,5 75% of congenital thoracolumbar curves will progress during growth, and 50% will require treatment. The factors associated with an unfavorable prognosis included early age at curve onset, vertebral anomaly type, and deformity location. The risk for progressive deformity was increased in the presence of block vertebrae, wedge vertebrae, hemivertebrae, two unilateral hemivertebrae, a unilateral unsegmented bar, or, most severely, a unilateral unsegmented bar associated with a contralateral hemivertebra at the same level. With each of these anomalies, the rate of deterioration was least severe in the upper thoracic region and most severe in the thoracolumbar region.
Early experience with atlantal hemirings and associated conditions (condylar aplasia or dysplasia, absent transverse ligaments, and C2 anomalies) seems to suggest that many of these patients present with craniocervical instability that may progress over time. It is common to intervene with a craniocervical fusion by the age of 4 years because the biomechanical stability of the occipitocervical complex fails over time.
29.7 Clinical Presentation and Evaluation
The clinical presentation of congenital vertebral anomalies is highly variable. Although pain is the most frequent presenting symptom among adults, incidental findings, association with other syndromes, and abnormal posture (e.g., torticollis) are the major pathways to diagnosis among children. Nevertheless, pain, weakness, sensory change, autonomic disturbance, and spine deformity may all exist alone or in combination as the presenting complaints. Pain may predominate in the midline or in a radicular fashion. Weakness and sensory changes can also be in a radicular pattern or can be part of a myelopathic syndrome involving a specific vertebral level. Autonomic disturbances, including bowel and bladder changes, may occur, with urinary incontinence often presenting early. Many patients with congenital vertebral body anomalies have symmetric fusions or blocked vertebrae and so present with no obvious spinal deformity. Indeed, if the fusion exists at one or two levels only, these individuals may not even be aware of its existence until it is revealed by radiographs taken for unrelated causes. Other individuals, however, can have much more obvious deformities that are detected at any time from birth to early adulthood.
The physical examination of a patient with suspected congenital vertebral anomaly should begin with general observations. A low-lying hairline or web neck deformity typical of Klippel-Feil syndrome or abnormalities of the ears or palate associated with Goldenhar syndrome may be noticed. Dwarfism without evidence of mental retardation may suggest achondroplasia, spondyloepiphyseal dysplasia, or some other type of skeletal dysplasia. Careful examination of the skin on the back is important to detect cutaneous stigmata of spinal dysraphism, including hairy patches, skin discoloration, dimples, lipomas, hemangiomas, and other findings. Examination of the extremities may reveal ligamentous laxity associated with Ehlers-Danlos syndrome or Larsen syndrome. Physical findings in the feet may include high arches or cavus deformity, which are common in Friedreich ataxia. The association of both foot and spine deformities strongly suggests spinal dysraphism or a generalized neuromuscular disorder. Limb length discrepancy is present if the patient is standing and the pelvis is out of the horizontal plane; this can be quantified by placing blocks of variable thickness under the short leg until the pelvis is level. The patient, dressed comfortably in an examination gown, should be standing during the examination of the spine, which should proceed in a cranial to caudal direction in a systematic fashion. In general, a “physiologic” curve is convex to the right, whereas a “pathologic” curve is convex to the left. Tenderness over the spine should be sought, as well as a determination of the range of motion. A complete neurologic examination should also be performed, with particular emphasis on the cranial nerves, sensorimotor function, and deep tendon reflexes. The patient’s gait and station may provide subtle clues for the presence of lower extremity or truncal weakness.
29.7.1 Cervical Region
Torticollis, a twisting of the neck with the head tilted toward the involved muscle and the chin rotated toward the opposite side (“cock robin” posture), is a common physical finding among children. Torticollis usually results from unilateral contracture and fibrosis of the sternocleidomastoid muscle; however, a retrospective study of 288 patients documented 53 (18.4%) with torticollis of nonmuscular causes.41 Although the majority of these had neurologic causes, almost a third were associated with vertebral anomalies. We routinely order plain cervical spine radiographs for children who present with torticollis to exclude vertebral anomalies before the initiation of physical therapy. In selected patients (e.g., those with vertebral anomalies or with severe deformity), thin-cut computed tomographic (CT) scans with two-dimensional reconstructions are ordered to further investigate the bony anatomy. Although torticollis usually responds to physical therapy, it must be carefully monitored because it may lead to severe, progressive problems.20
Although the majority of congenital vertebral anomalies in children are asymptomatic, several anomalies should be considered when children present with neck pain or torticollis. Atlantal hemirings and congenital upper cervical anomalies are relatively frequent causes of torticollis in younger children. Cervical ribs, or anomalous ribs in the cervical region that point downward, vary in size from tiny ossicles to fully formed ribs. The ribs are usually asymptomatic unless they are large enough to compress nerves or vessels. Symptoms of venous compression include pain in the ulnar distribution, neck pain, and pain along the involved part of the brachial plexus.42 Another uncommon anomaly of the cervical spine is an absent pedicle, which is also largely asymptomatic. In 1990, a review of 55 patients found that 31 had cervical pain, most often after trauma. The typical radiographic triad is (1) the false appearance of an enlarged neural foramen caused by the absent pedicle, (2) a dorsally displaced ipsilateral articular mass and lamina with a dysplastic and reversed facet joint, and (3) a dysplastic ipsilateral transverse process.43
29.7.2 Thoracolumbar Region
Except after trauma, neurologic dysfunction associated with spinal deformity of the thoracic level is usually insidious in onset and slow in progression. Most cases are associated with idiopathic or congenital spinal deformities, particularly kyphosis. The terms kyphosis, lordosis, and scoliosis refer to abnormally increased convexity in the curvature of the thoracic spine (lateral view), increased anterior concavity in the curvature of the cervical and lumbar spine (lateral view), and increased lateral deviation from the normally straight vertical line of the spine (posterior view), respectively.
Scoliosis of the thoracic and lumbar spine usually presents as a painless deformity and has commonly been found in school screening programs. Juvenile idiopathic scoliosis is defined as scoliosis detected in children between 3 and 10 years of age, and adolescent idiopathic scoliosis occurs between 10 and 18 years of age. The prevalence appears to decrease with increasing curvature: 2 to 3% for a curve magnitude of more than 10 degrees, 0.3 to 0.5% for a curve of more than 20 degrees, and 0.2 to 0.3% for a curve of more than 30 degrees. Progression appears to increase with increasing curvature.26
The most sensitive diagnostic screening test for thoracolumbar spinal deformity is the forward-bending test. As the patient bends forward, the presence of a rib prominence or rotation is highly suggestive of an underlying curve and can be measured with a horizontal inclinometer. A plumb line dropped from the center of the occiput to the gluteal cleft is able to detect any lateral deviation of the trunk. Scoliotic curves should be described by their apex and location, such as a right thoracic curve, left lumbar curve, and so on. Sagittal plane deformity should also be assessed, with an evaluation for excessive kyphosis or lordosis.
29.7.3 Lumbosacral Region
Segmentation anomalies are frequently found in the lumbar and sacral spine, but they rarely produce neurologic symptoms or signs in children. Hemiblock and wedge vertebrae contribute to scoliotic and kyphotic deformities.20
Caudal agenesis encompasses numerous congenital malformations of the lumbosacral spine ranging from simple anal atresia to the absence of sacral, lumbar, and possibly thoracic vertebrae to the most severe segmentation failure of the lower extremities, sirenomelia.6,20 Children with caudal agenesis generally show normal cognitive development but are often paraplegic and seldom have associated treatable neurologic conditions, such as spinal stenosis and tethered cord syndrome. Caudal agenesis is discussed in more detail in a subsequent section.
29.8 Diagnostic Evaluation
29.8.1 Plain Radiographs and Curve Measurement
Plain radiographs form the foundation from which congenital vertebral anomalies are detected and treated. Cervical spine radiographs—including lateral, anteroposterior, and odontoid views—are mandatory when clinical evidence of cervical abnormalities is present. Careful interpretation, however, is paramount because normal vertebral growth and ossification may show variable patterns. In most patients with congenital vertebral anomalies of the cervical spine, dynamic flexion and extension studies should be performed to determine whether instability is present and to assess whether there is any change in the spinal canal width. If thoracolumbar scoliosis is suspected, then upright lateral and anteroposterior plain radiographs of the entire spine should be obtained for initial curve assessment. The Cobb method is recommended by the Scoliosis Research Society to measure the degree of spinal deformity. It is applicable to both idiopathic and congenital curves. To use the Cobb method, the examiner chooses the most tilted vertebrae above and below the apex of the curve. Lines are then drawn extending from the superior end plate of the top vertebraand the inferior end plate of the bottom vertebra. The angle formed when intersecting lines are drawn perpendicular to the above lines is the Cobb angle. Imaging the entire spine on a single radiograph allows an assessment of the magnitude of the curve, the pattern of vertebral anomalies, and the overall balance in sagittal and coronal planes. Equally important, it also allows an assessment of progressive decompensation and spinal balance.
29.8.2 Special Imaging
CT provides detailed information regarding the bony anatomy of congenital vertebral anomalies and is invaluable in the treatment-planning phase of these disorders. Thin-cut (1 mm) axial CT scans with multiplanar computer reformatting in the sagittal and coronal planes (1 mm) give further insight into the anatomy of the anomaly, especially those in the cervical region. In addition, two-dimensional parasagittal reconstruction of the atlantoaxial region is critical when the use of C1–2 instrumentation is being contemplated for the treatment of instability at the craniocervical junction.44–46 With the advent of magnetic resonance (MR) imaging, the use of CT myelography for the evaluation of spinal cord anatomy has decreased precipitously. There are cases, however, where CT myelography may be preferable to MR imaging, such as when spinal instrumentation overlies the area of interest and produces significant MR imaging artifact.
MR imaging is the study of choice for evaluating spinal cord pathology. Congenital anomalies and acquired defects of the spinal cord can be identified at all spinal levels. Because of the inherent difficulty of detecting bony abnormalities with MR imaging, however, more than one imaging modality is often required. Plain radiographs, CT, and MR imaging all may be necessary to completely understand the anatomy of a given lesion. Because multiple spinal anomalies are commonly present in the individual patient, it is important to image the entire spine if any malformation is suspected.
In infants, ultrasound can be useful initially because monitoring devices do not need to be removed, transport is often not required, and patients can be closely observed during the examination. Furthermore, ionizing radiation is not involved, sedation is not required, and most institutions have the requisite equipment.47 Among other things, ultrasound can provide information about posterior cystic lesions, Chiari malformation, syringomyelia, diastematomyelia, and the position of the conus.48
29.9 Associated Anomalies
The physical proximity of the branchial arches, genitourinary system, viscera, and other tissues to the spinal column during development permits a localized teratogenic event to have an effect on multiple organ systems. The branchial arches arise from the intermediate mesoderm adjacent to the cervicothoracic somites. Thus, derangement of the somites or developing vertebrae can be associated with malformations of the structures that arise from the branchial arches. These structures include the outer ear, ossicles, semicircular canals, mandible, and parts of the maxilla and hyoid bone.49,50 Patients with Apert, Crouzon, or Treacher Collins syndrome, for example, tend to have craniocervical abnormalities or blocked cervical vertebrae and associated complex craniofacial anomalies. Those with Goldenhar syndrome often have cervicothoracic scoliosis associated with a hemifacial microsomia, with severe ear deformities frequently noted. The unilateral hypoplasia or absence of the ear in this syndrome is associated with mandibular hypoplasia, macrostomia, an ocular dermoid, and fused vertebrae or hemivertebrae, usually in the lower cervical and upper thoracic levels.50
The pronephros also originates at the level of the lower cervical spine, and teratogenic factors at those levels can be associated with abnormal development of the genitourinary system.51 Genitourinary anomalies, including unilateral kidney, ureteral obstruction, and duplication of the kidney, may occur in up to 25% of patients with congenital scoliosis.52
Because of the close proximity of the developing spinal cord to the osseous structures in the spine, malformations of the central nervous system often coexist with congenital vertebral anomalies.53 For example, Bradford and colleagues54 found a 38% incidence of intraspinal anomalies in 42 patients with congenital scoliosis. There was also a 52% incidence of spinal cord abnormalities, with unilateral unsegmented bars and contralateral hemivertebrae. Other studies have shown that split-cord malformations are present in 5 to 20% of patients with congenital scoliosis.55
29.10 Congenital Vertebral Anomaly Syndromes
29.10.1 Klippel-Feil Syndrome
Klippel-Feil syndrome is a relatively rare entity that is classically described as the clinical and radiographic triad of short neck, low posterior hairline, and cervical vertebral segmentation abnormalities. All three components of the triad occur in only about 50% of patients with the syndrome, leading to the alternative description of a Klippel-Feil “variant,” which encompasses a larger number of patients. Although the true incidence of Klippel-Feil syndrome or variant in the general population is unknown, it is estimated in reports to be 1 in 42,000 live births, or somewhere between 0.02 and 0.7% of the population.56–59
Klippel-Feil syndrome is usually diagnosed in children after incidental radiographic studies. Gross spinal deformities, torticollis, scoliosis, or acute cervical symptoms occurring immediately after a relatively mild injury, however, may indicate the syndrome before imaging is obtained. In a minority of cases, Klippel-Feil syndrome presents as chronic cervical symptoms lasting months: progressive neck and upper extremity pain, paresthesia, or weakness; ataxia; vertigo; headaches; and vision problems.
Klippel-Feil syndrome is most often seen as a sporadic disorder, although autosomal-dominant and autosomal-recessive cases have been reported. Mutations in the GDF6 and GDF3 genes, both encoding bone and cartilage regulatory proteins, have been identified to cause the disease. Several different hypotheses have been proposed over the years to explain its origin and include primary vascular disruption, global fetal insult, primary neural tube abnormality, genetic predisposition, and failure of facet joint segmentation (see the previous section on etiology).29,32,34,60
Abnormalities in virtually every organ system have been associated with Klippel-Feil syndrome. These include Chiari 1 malformation with or without syringohydromyelia and developmental anomalies of the head such as high-arch palate, lid ptosis, cleft palate, facial nerve palsies, Duane contracture of the lateral rectus muscle, and mixed-type hearing loss.61,62 Between 30 and 60% of patients with Klippel-Feil syndrome have genitourinary abnormalities, including unilateral kidney, malrotation of a normal kidney, ectopic kidney, horseshoe kidney, and renal pelvic and ureteral duplication, or genital anomalies.61,63,64 Unilateral renal agenesis, the most common anomaly among patients with Klippel-Feil syndrome, is 400 times more common in these patients than in the unaffected population. Among cardiovascular anomalies (which affect 4 to 5% of patients with Klippel-Feil syndrome), the most commonly reported are coarctation of the aorta, patent ductus arteriosus, mitral valve disease, and ventricular septal defects. Pulmonary anomalies include failure of lobe development, rib fusion, and ectopic lung secondary to a deformed trunk and rib cage. Syndactyly, elevation of the scapula (Sprengel deformity), and pterygium colli (webbing of the skin on the lateral side of the neck) have been reported.61,65 Hearing loss is common in patients with Klippel-Feil syndrome, occurring in up to 30%.66 Accessory cervical ribs are found in 15% of patients with Klippel-Feil syndrome, whereas only 1% of individuals in the general population are similarly affected.65
The Klippel-Feil syndrome involves segmentation anomalies of the spine, including fused vertebrae, hemivertebrae, occipitalization of the atlas, scoliosis, and spina bifida occulta (▶ Fig. 29.2). This places the cervical spinal cord at risk primarily because of the resulting biomechanical abnormalities. A common abnormality is congenital fusion of the second and third cervical vertebrae in conjunction with assimilation of the atlas.67 Although this combination has been reported to result in atlantoaxial subluxation approximately 50% of the time,68 our institutional experience suggests a much lower rate of instability (about 5%). Furthermore, congenital fusions over several spinal segments reduce the ability of the cervical spine to compensate for excessive forces in flexion, extension, rotation, and lateral bending, and stresses that might be tolerated by a patient who has a normal neck are not safe for one who has a cervical spine with limited mobility. Forces applied to the neck develop moments at each motion segment, and if only one or two motion segments exist where there should be eight, the force may exceed the strength of the restraining ligaments, leading to dislocation and injury to the spinal cord. In reported cases of patients with Klippel-Feil syndrome and spinal cord injury, the trauma is often minor or indirect, with the dislocation frequently self-reducing.69,70 Congenital spinal stenosis may also be present in patients with Klippel-Feil syndrome, increasing the risk that even minor stress applied to the cervical spine can lead to neurologic injury.71,72 Anesthetic considerations for these patients are significant because children with Klippel-Feil syndrome are at high risk for spinal cord injury during laryngoscopy, intubation, and positioning.73,74
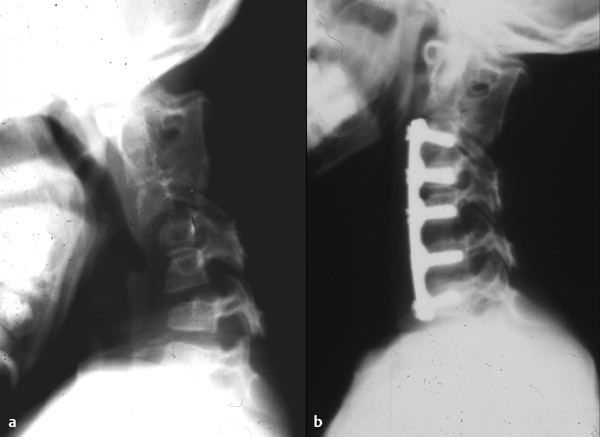
Fig. 29.2 A 9-year-old girl presented with Klippel-Feil syndrome. (a) Plain lateral cervical spine radiograph in flexion reveals significant subluxation at the C4–5 and C6–7 levels. (b) Postoperative lateral radiograph demonstrates multilevel anterior cervical diskectomy and fusion with plating. The patient went on to have a successful fusion.




