Craniofacial Syndromes
The craniofacial syndromes are a heterogeneous group of rare conditions in which premature suture closure (craniosynostosis) occurs alongside other manifestations of disordered craniofacial development,1 as well as additional skeletal abnormalities that include, in particular, those of the hands and feet.2,3
The conditions seen most frequently are the eponymous syndromes of Crouzon, Apert, Pfeiffer, Saethre-Chotzen, Muenke (FGFR3-associated synostosis), and Carpenter, as well as craniofrontonasal dysplasia. Less commonly encountered are Antley-Bixler,4,5 Jackson-Weiss,6 and Boston-type syndromes.
The aim of care is to ensure that affected children realize their full developmental potential. Although a major component of treatment is surgical, there is now greater recognition of the needs of the child as a whole. The centers best equipped to achieve this are those that can field a multidisciplinary team; in addition to neurosurgeons, plastic surgeons, and maxillofacial surgeons, such teams include specialists in otorhinolaryngology (ear–nose–throat) and audiology, genetics, pediatrics, neurology, psychology, orthodontics, respiratory medicine, speech therapy, ophthalmology, and, of course, pediatrics—all experienced in the management of the complex problems these children so frequently have.7
After a brief account of the syndromes and their genetic basis, this chapter describes first the principles of management and then the complications that affected children are at particular risk for. It ends with a description of the surgical procedures most commonly deployed in the treatment of children with these complex problems.
20.1 The Syndromes
It was hoped that when the genetic basis for many of the craniofacial syndromes was discovered in the 1990s that their classification would change from the one based on the eponymous nomenclature that had been in use for so many years. Unfortunately, the situation proved to be more complex, as the genetic “overlap” between Crouzon syndrome and Pfeiffer syndrome demonstrates—a single mutation is capable of causing both Pfeiffer syndrome and Crouzon syndrome, and mutations on either of two chromosomes are responsible for Pfeiffer syndrome.8–10
It therefore remains important for clinicians to recognize the typical features of each syndrome as traditionally described as well as be conversant with the underlying genetic mutations when these are known.
20.1.1 Crouzon Syndrome
Crouzon described the syndrome that bears his name in 1912. One of the more frequently encountered craniosynostosis syndromes,11 it is transmitted as an autosomal-dominant condition (as with Apert syndrome, there is a significant influence of increased paternal age),12 although it occurs with nearly equal frequency as a new mutation.
Typical features include a retruded maxilla that leaves the lower teeth projecting anterior to the upper teeth (class 3 malocclusion), a “beaky” nose, a recessed frontal region (brachycephaly) due to bicoronal synostosis, and prominent eyes (exorbitism) due to the combined recession of the infra- and supraorbital regions (▶ Fig. 20.1).
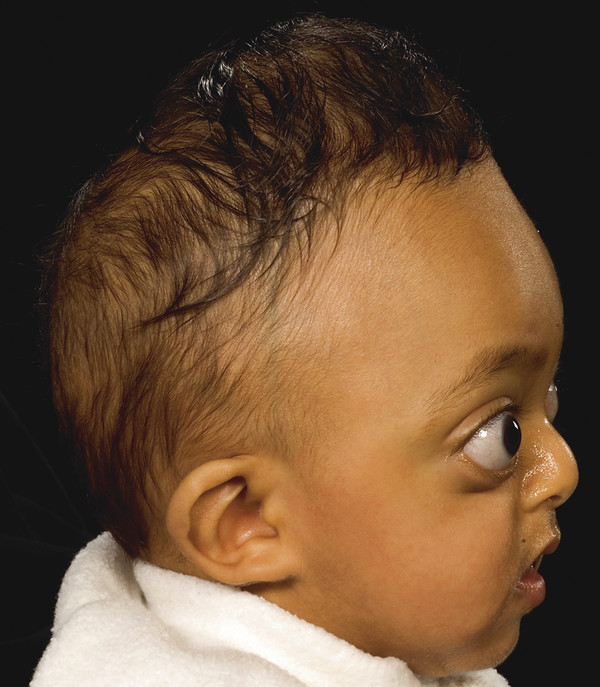
Fig. 20.1 Crouzon syndrome.
Its expression is highly variable, ranging from severe exorbitism with midface retrusion and airway obstruction at one extreme to a mild prominence of the eyes at the other. The degree of craniosynostosis also varies, although the coronal sutures are most commonly affected. A catch for the unwary is that suture fusions need not be present at birth but may appear during the first 2 years of life,13 with a high risk for the development of raised intracranial pressure (ICP).14
Extracranial manifestations15 seen in severe cases include various types of predominantly cervical vertebral fusion16,17 and ankylosis affecting particularly the elbows.17
A genetically distinct type of Crouzon syndrome is associated with rugated, thickened skin and hyperpigmentation affecting particularly the flexure creases—acanthosis nigricans.18
Intelligence may be normal in children with Crouzon syndrome, but the more severe the phenotype, the more likely the child is to have developmental and learning difficulties. Marked intellectual compromise was present in 3% in the series of Kreiborg.19
20.1.2 Apert Syndrome
Apert described the rare20 condition that now bears his name in 1906.
The affected child has a head that is tall and shortened from front to back (turribrachycephaly) combined with midfacial (maxillary) retrusion, proptosis, a downward cant to the palpebral fissures, and hypertelorism21 (▶ Fig. 20.2a). The essential clinical feature, however, is complex fusion (syndactyly) of the fingers and toes22–24 that may require frequent surgical procedures before functional effectiveness is achieved25 (▶ Fig. 20.2b). Visceral26 and cutaneous27 abnormalities can also occur.
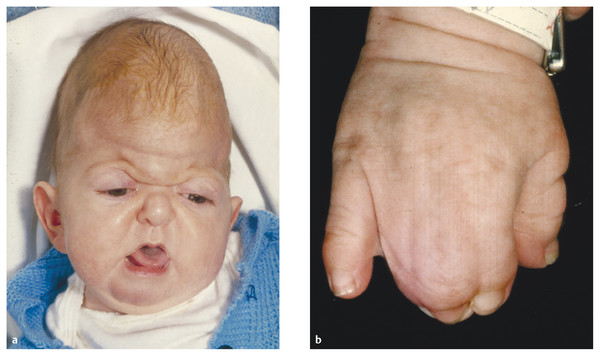
Fig. 20.2 Apert syndrome. Typical facial (a) and hand (b) appearances.
At birth, the child with Apert syndrome may have fusion of only the coronal sutures, while the sagittal and (in particular) the metopic sutures are widely open. Fusion of all sutures, however, is progressive and usually complete before the age of 2 years.
Palatal abnormalities28 ranging in severity from frank clefts to bifid uvula are common and occur with a frequency of up to 75%.29,30
Cervical vertebral fusions that, like the suture fusions, are often progressive occur in more than half of affected children, although it is unusual for them to become clinically significant.31
Developmental and learning difficulties are the norm, although a combination of developmental assessments designed for non-Apert children with low societal expectations may overestimate their severity.32 Although a small percentage of children may complete secondary education (usually only with assistance), many drop out of mainstream education during their primary school years, and a small percentage are too severely affected to participate in the mainstream education system at anything above kindergarten level.33,34 The long-term study of Allam et al of the treatment of Apert syndrome includes a description of the eventual social integration of these children.35
20.1.3 Pfeiffer Syndrome
Pfeiffer syndrome is an autosomal-dominant craniosynostosis-associated syndrome characterized by suture fusions that range from bicoronal synostosis alone to pan-synostosis (with or without the cloverleaf skull deformity; see below).36 Those affected have digital abnormalities,3 such as curved and shortened thumbs and great toes37 (▶ Fig. 20.3), and (less commonly) digital fusions (but of a lesser degree than those seen in Apert syndrome3).
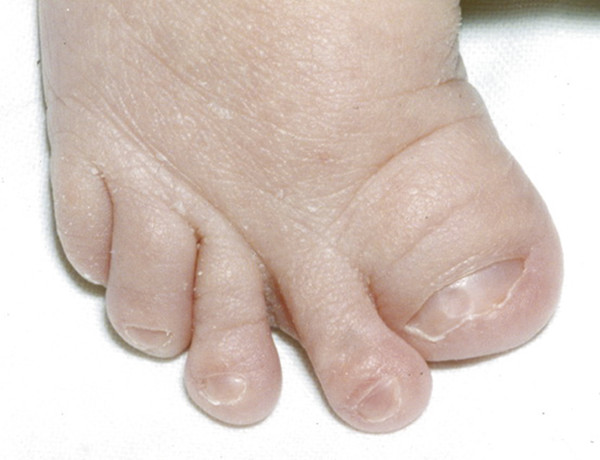
Fig. 20.3 Pfeiffer syndrome. Typical hallux deformity.
Cohen38 divided Pfeiffer syndrome into three types based on clinical severity. Children with type 1, those least affected, may display little more than bicoronal synostosis and midface retrusion (in addition to their digital abnormalities) (▶ Fig. 20.4). Their neurocognitive development may be unaffected, particularly if early complications have been aggressively treated.39
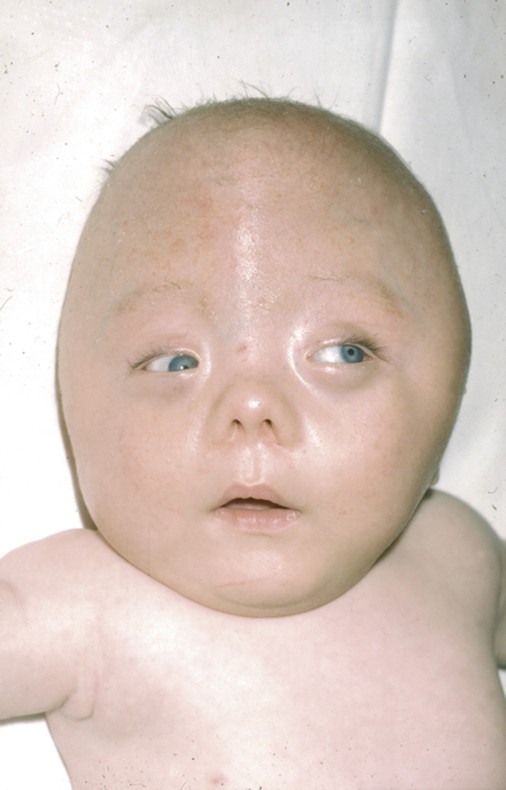
Fig. 20.4 Pfeiffer syndrome type 1.
In types 2 and 3 Pfeiffer syndrome, the degree of midface and frontal retrusion is severe enough to obstruct the upper airway and cause ocular protrusion sufficient to threaten the corneas (▶ Fig. 20.5). The shortening of the skull base and crowding of the posterior fossa due to the bilateral lambdoid component of the pan-synostosis produces an increased risk for hydrocephalus. Ankylosis (bony and soft tissue) of the elbows17 and knees is common, as are fusions of the cervical vertebrae.40
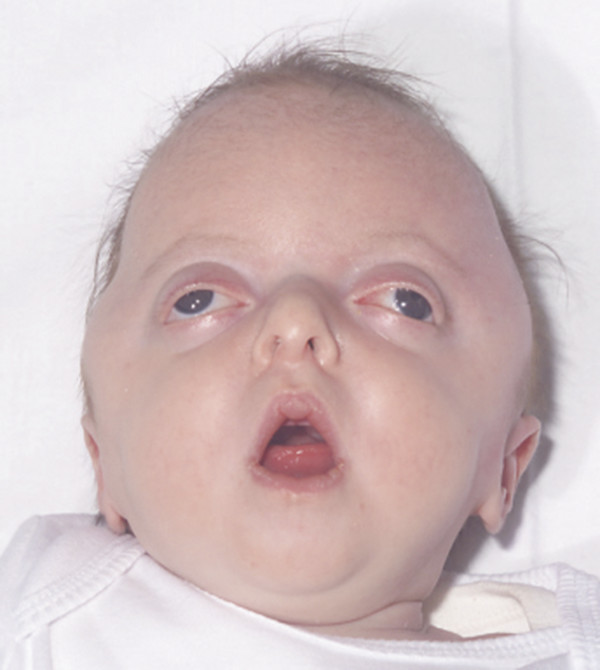
Fig. 20.5 Pfeiffer syndrome type 3.
The difference between types 2 and 3 is that type 2 has the cloverleaf pattern of skull deformity (see below).
Neurocognitive development in types 2 and 3 Pfeiffer syndrome is usually delayed, although with active intervention aimed at improving the airway and reducing raised ICP, the outlook is not as dire as was once assumed.41
20.1.4 Cloverleaf Skull (Kleeblattschaedel) Deformity
Kleeblattschaedel anomaly, or cloverleaf skull, is the descriptive term given to a particularly severe form of synostosis-associated cranial deformity,42 one that poses a particular challenge for the craniofacial surgeon.43,44
Although it usually occurs in association with Pfeiffer syndrome (of which it forms type 2), it can occasionally complicate Apert and Crouzon syndromes.
Cloverleaf skull is produced by a particular combination of suture fusions and raised ICP due to hydrocephalus. The sagittal and squamoparietal sutures are open, but a complex form of bicoronal synostosis results in a bony constriction band that runs posteriorly from the pterions to the lambdoids. When hydrocephalus is added to this bony pattern, the infant’s skull expands upward (above) and laterally (below) the constriction band to produce a characteristic trefoil (cloverleaf) shape (▶ Fig. 20.6).38,45–47
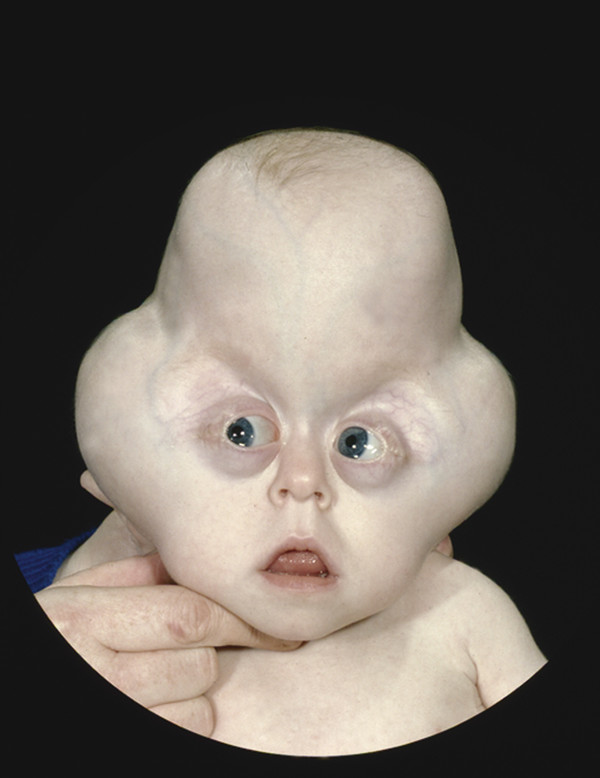
Fig. 20.6 Pfeiffer syndrome type 2. Cloverleaf pattern.
20.1.5 Saethre-Chotzen Syndrome
Saethre in 1931 and Chotzen in 1932 described an autosomal-dominant condition in which are combined with great variability48 features of coronal synostosis (uni- or bilateral), digital abnormalities that include short digits and partial syndactyly,49 a low frontal hairline, a prominent nose, ptosis (▶ Fig. 20.7), and more rarely fusions of the cervical spine.50
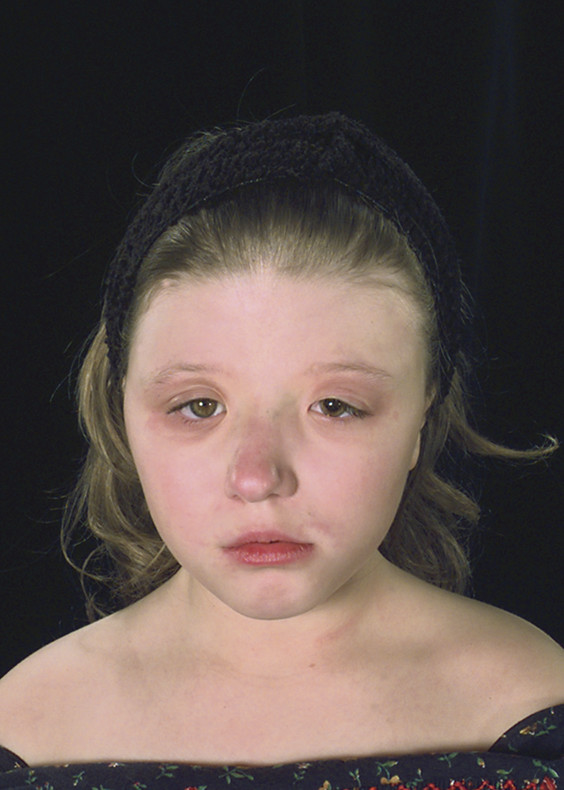
Fig. 20.7 Saethre-Chotzen syndrome.
Complications like exorbitism and airway obstruction are uncommon, raised ICP is rarely of functional significance,51 and the neurocognitive outcome may be only modestly affected, if at all.48
20.1.6 Muenke (FGFR3 Mutation) Syndrome
This condition, one of the less severe of the craniofacial syndromes, is of interest because rather than being first described on the basis of the appearance of those affected, it was “discovered” during the explosion of knowledge about the genetic basis of the craniofacial syndromes that occurred during the 1990s.52
Muenke syndrome has many manifestations,53 but the synostosis typically affects either one or both coronal sutures.54 Indeed, many patients previously diagnosed with isolated unicoronal craniosynostosis are now known to carry the FGFR3 mutation.55 Those with bicoronal synostosis typically have a broad and shallow supraorbital region with a protruding upper forehead (▶ Fig. 20.8).
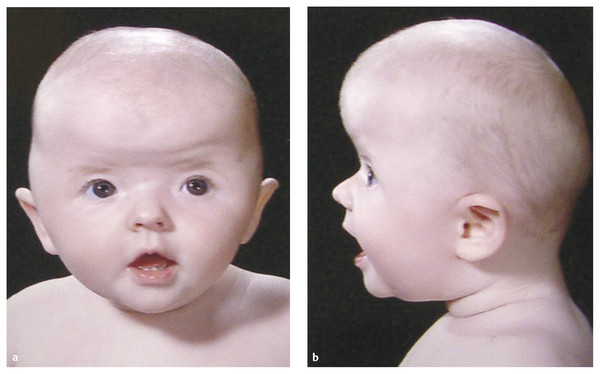
Fig. 20.8 (a,b)FGFR3 mutation (Muenke syndrome).
Complications like raised ICP and airway obstruction are rare; however, although the development of a child with Muenke syndrome may be unaffected, a degree of learning difficulty is not uncommon.52
20.1.7 Craniofrontonasal Dysplasia
In this X-linked56 syndrome (▶ Fig. 20.9), bicoronal synostosis (usually asymmetric in its effect) is associated with hypertelorism, wiry (“unruly”) hair, a prominent gap between the central incisors, a bifid nose,57–59 and sometimes abnormalities of the optic discs.60 Development is usually unaffected, and treatment is indicated predominantly for cosmetic reasons.61
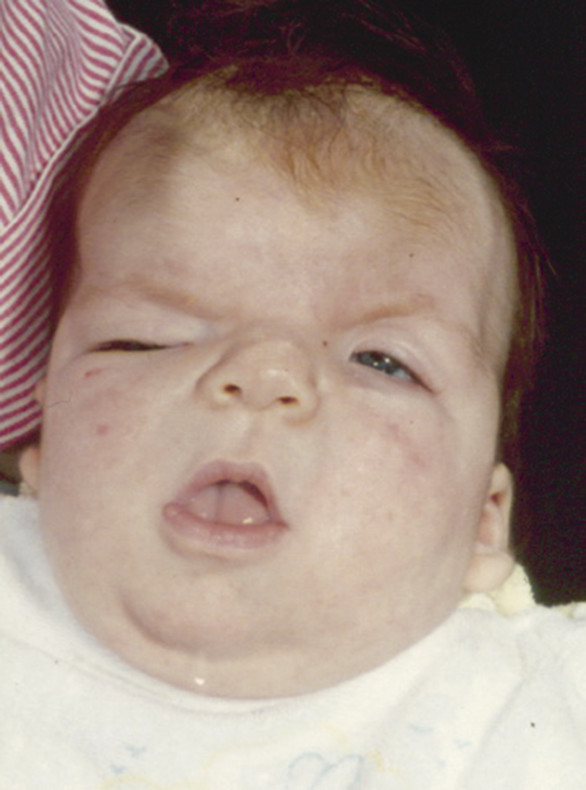
Fig. 20.9 Craniofrontonasal dysplasia.
20.1.8 Carpenter Syndrome
This very rare condition62 is mentioned here for completeness because it is the only one of the craniofacial syndromes that has an autosomal-recessive pattern of inheritance. Also known as acrocephalopolysyndactyly because of the extra digits that form part of the clinical picture, the cranial deformity is due to various combinations of craniosynostosis.63,64
20.2 The Molecular Genetics of Syndromic Craniosynostosis
With the exception of Carpenter syndrome (autosomal-recessive65) and craniofrontonasal dysplasia (X-linked56), the craniosynostosis-associated syndromes have an autosomal-dominant pattern of transmission. (For a summary linking the clinical features of syndromic craniosynostosis to their underlying genetic abnormalities, see the review by Rice.1)
The realization that many of the craniofacial syndromes are monogenetic (due to the mutation of a single gene) plus the investigation of families with several affected members led in the 1990s to an intense examination of the genes for a fibroblast growth factor group of tyrosine kinase receptors (FGFR I, FGFR II, and FGFR III) as candidate genes. These receptors are well preserved across a range of species and are involved (among many other activities) in cranial and limb development.66–69 It is now known that a particular position within each FGFR protein is strongly linked to craniosynostosis because mutations in their genes (FGFR1, FGFR2, and FGFR3) cause Pfeiffer, Apert, and Muenke syndromes, respectively.70
Tyrosine kinase receptors straddle the cell membrane and have extracellular, transmembranous, and intracellular components. Mutations responsible for many of the craniosynostosis syndromes affect the extracellular component of FGFR II and act by provoking “gain of function”—the receptor overresponds either to its (appropriate) fibroblast growth factor (FGF) or to other (inappropriate) chemical signals.10
In contrast, mutation of the transcription factor TWIST gene (which is part of the FGFR signaling pathway and like the FGFs is expressed in calvarial bone71), which is responsible for Saethre-Chotzen syndrome, acts by causing functional loss.72
At a molecular level, the situation is complicated by the functional diversity of the various FGFR mutations.68 For example, there are a number of individual variations of (say) FGFR273 responsible for what from a clinical perspective would be labeled Crouzon syndrome—approximately 30 have been identified to date. However, 95% of children with Apert syndrome have one of only two genetic variations on the extracellular portion of FGFR II.8,18,29,74 As already described, not only may Pfeiffer syndrome be associated with mutations in either FGFR2 or FGFR1,9,75 but identical mutations in FGFR2 can cause both Pfeiffer and Crouzon syndromes (▶ Table 20.1).8
| GENE (CHROMOSOME) | RAB23 (6) | TWIST (7) | FGFR1 (8) | FGFR2 (10) | FGFR3 (4) | |
| SYNDROME | References | |||||
| Apert | ✓ | 74 | ||||
| Crouzon | ✓ | 8 , 174 , 175 | ||||
| Crouzon with acanthosis nigricans | ✓ | 18 , 75 | ||||
| Pfeiffer | ✓ | ✓ | 8 , 9 , 75 , 176 , 177 | |||
| Pfeiffer and Crouzon | ✓ | 8 | ||||
| Saethre-Chotzen | ✓ | 48 | ||||
| Muenke | ✓ | 52 | ||||
| Carpenter | ✓ | 65 |
Although confirmation of a particular mutation may not affect a child’s immediate management, it does allow a more informed prognosis to be given to parents. Furthermore, the discovery of the FGFR3 mutation in a child previously thought to have nonsyndromic unisutural synostosis, for example, will warn the craniofacial surgeon that a degree of relapse/reversion may occur following reconstructive surgery76 (see below).
Knowledge of the responsible mutation also has important implications for genetic counseling. Parents who already have an affected child may wish to avail themselves of the opportunity not only for prenatal ultrasound examination of the fetus77–80 but also for preimplantation diagnosis when considering further pregnancies.81,82
Research continues into the possibility of either gene substitution therapy following fetal diagnosis or a “vaccine” that interferes with FGFR function and could block some or all of a syndrome’s most disabling features.83
20.3 Principles of Surgical Management
It is essential that children with a craniofacial syndrome be referred to a suitably staffed unit as early as possible so that the correct diagnosis (both genetic and clinical) can be made, the risk for complications assessed, and a management plan tailored to each individual child’s needs devised.
As the majority of craniofacial syndromes result from a mutation of a particular gene (usually of the FGFR series), it should be no surprise that such mutations continue to exert their ill effects for as long as the cranial and facial skeleton is growing. It is this phenomenon that causes the anomalies corrected during reconstructive surgery to eventually drift toward their preoperative state despite what appeared at the time of surgery to have been a satisfactory result, even to the extent that repeated surgery is required.84,85 Wong et al wrote of their results of fronto-orbital surgery: “In the final analysis, the expression of the underlying genetic defect probably is the major determinant of the final fronto-orbital position despite our best surgical efforts.”86 Or, as Kohan et al concluded in their report of twins with Pfeiffer syndrome (after each twin had received a different treatment strategy), “The genetic mutation may have overridden the different surgical interventions.”39
The degree to which such reversion occurs (which is not to be confused with relapse due to the failure of bone grafts or plates and screws or any injurious effects of the procedure itself) is influenced by the severity of the gene’s phenotypic expression and the age of the child when surgery is performed. Thus, reversion is seen most often in the severely affected child operated upon at a young age whose craniofacial growth is proceeding rapidly; it is less of an issue in a more mildly affected child whose growth is nearly complete87 (and, of course, in those whose bicoronal synostosis (say) turns out to be nonsyndromic88).
All this has important implications for the timing of surgery. If the result of a reconstructive procedure for a child with syndromic synostosis is to be stable into adulthood, it should ideally be postponed until the most active growth phase of the area being operated upon (e.g., the fronto-orbital region or the maxilla) has been completed unless a particular functional complication demands earlier intervention.89–91 This principle also holds good for the management of hypertelorism.92
In practice, this means that the majority of children with Apert syndrome93 and the more severe forms of Pfeiffer and Crouzon93 and cloverleaf94 syndromes may require several procedures during their early years to treat such functional issues as raised ICP, exorbitism, airway obstruction, and psychological issues related to their appearance (e.g., teasing).35 In contrast, for more mildly affected children (e.g., with Saethre-Chotzen or Muenke syndrome76), reversion is less of an issue, even after early surgery.
In brief, the complexity of decision making for children with syndromic craniosynostosis means that if they are to achieve their optimal developmental outcome, their management should be undertaken only in multidisciplinary units specializing in their care.51,91
20.4 Functional Complications of Syndromic Craniosynostosis
20.4.1 Raised Intracranial Pressure
Raised ICP is a well-recognized complication of syndromic craniosynostosis.95,96 Its incidence is strongly related to the severity of the phenotype; raised ICP is unusual in Muenke and Saethre-Chotzen syndromes and nearly inevitable in Pfeiffer type 2 syndrome (cloverleaf skull deformity).
In a study of 49 children with Crouzon syndrome from our unit,97 raised ICP occurred in 30. The predominant cause was craniocerebral disproportion in 17, hydrocephalus in 10, and airway obstruction in 3. It recurred in 14 of the 30 after successful treatment of the first episode, and 3 experienced a third episode. Those in whom raised ICP was first diagnosed before the age of 1 year were those in whom it was most likely to recur, although not necessarily for the same reason (e.g., the onset of hydrocephalus following a vault expansion for venous hypertension).
Marucci et al98 studied our cases of Apert syndrome. Raised ICP first occurred at an average age of 18 months in 20 of 24 children. It was treated with vault-expanding surgery in 16, cerebrospinal fluid (CSF) diversion in 2, and relief of airway obstruction in 2. It recurred in 7 patients, on average 3 years and 4 months later.
Our experience of Pfeiffer syndrome shows a similar incidence of raised ICP requiring intervention as that in our children with Apert syndrome.99
Raised ICP can be responsible for a progressive deterioration in vision that leads eventually to blindness.100,101 In one assessment of visual acuity in children with syndromic craniosynostosis, 40% had function in their better eye below a level (6/12) that would allow them to hold a driving license in the United Kingdom.102
Whether raised ICP in syndromic craniosynostosis (in the absence of hydrocephalus) can affect cognitive development over and above the effects of other frequent complications, such as chronic airway obstruction, feeding difficulties, and failure to thrive, plus any direct effects of the mutated gene upon the brain itself, is debatable. Renier et al,34,95 as well as other investigators, have proposed such a connection, but it is always difficult to untangle the effects of raised ICP alone from those of other manifestations of a child’s phenotype (e.g., how severely their breathing is affected). However, until more definite information becomes available, the neurosurgeon’s default position must always be to assume that raised ICP left untreated can indeed impair developmental outcome.
The presence of increased ICP is suggested by:
Clinically: bulging of still-open fontanels; stretched sutures and craniectomy defects; in older children, headache and vomiting
Radiologically: a generalized “beaten-copper” appearance on skull radiographs in the presence of multiple suture fusions103; progressive ventriculomegaly and effacement of the cortical sulci on magnetic resonance (MR) images and/or computed tomographic (CT) scans104
Ophthalmologically: papilledema105 with or without abnormal electrodiagnostic studies and a decrease in visual acuity106–108
On invasive ICP monitoring, the “gold standard” for the assessment of ICP:95,96 It is important to recognize that the figures most often used to interpret the results of ICP monitoring in childhood (normal, < 10 mm Hg); borderline, 11 to 15 mm Hg; raised, > 15 mm Hg) are “best guesses” based on a variety of assumptions. (Understandably, the parents of healthy children are reluctant to subject them to an invasive procedure performed for essentially academic interest.)109 Indeed, whether an ICP of 15 to 20 mm Hg can be responsible (in the absence of other problems) for any functional impairment is debatable.
It is important to remember that although the ICP may be normal when a child first presents, the dynamic nature of the syndromic craniosynostosis process means that ICP monitoring (which in our unit includes regular ophthalmic assessments) should continue until a child is at least 8 years old; our experience is that it is unusual for raised ICP to develop or recur at an age older than that.
Causes of Raised Intracranial Pressure in Syndromic Craniosynostosis
Although the four principal causes of raised ICP in children with syndromic craniosynostosis are considered separately here, some overlap is common. Indeed, they often interact to result in a vicious cycle (▶ Fig. 20.10); during active (rapid eye movement, or REM) sleep, airway obstruction and periods of apnea cause CO2 retention, drops in SpO2, and cerebral vasodilation that produce waves of very high pressure lasting 10 to 20 minutes superimposed upon a baseline that may be only moderately elevated (▶ Fig. 20.11). During these waves of high pressure (most obvious on overnight recordings), the cerebral perfusion pressure can fall to as low as 14 mm Hg.110
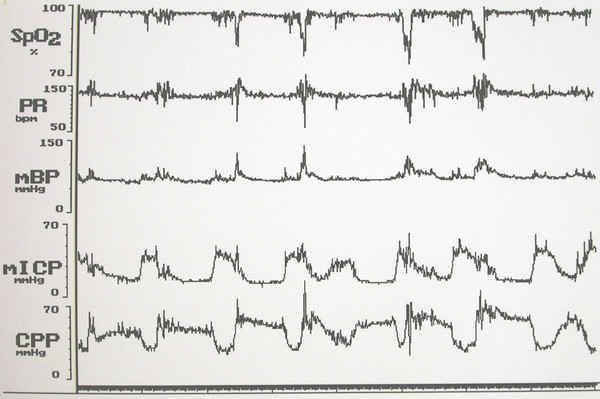
Fig. 20.10 Overnight record showing waves of elevated intracranial pressure associated with drops in SpO2, elevations in blood pressure, fluctuations in pulse rate, and drops in cerebral perfusion pressure.
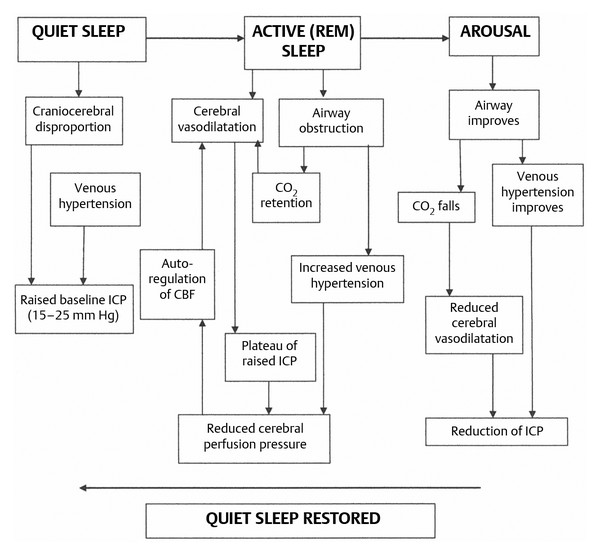
Fig. 20.11 The vicious cycle of raised intracranial pressure/airway obstruction/venous hypertension. CBF, cerebral blood flow; ICP, intracranial pressure; REM, rapid eye movement.
Craniocerebral Disproportion
Intracranial volume (ICV) can conveniently be measured with CT data111 in children with112 (and without113) craniosynostosis, but it has proved an unreliable predictor of raised ICP.114,115
Although it was once thought that the cause of raised ICP in syndromic synostosis was restraint of the growing brain by a skull that could not adequately accommodate it, it is now recognized that this is a relatively unusual situation. Indeed, in Apert syndrome, the ICV, although normal at birth, may actually be greater than normal by the time raised ICP appears.113,116 Fortunately, the various forms of vault expansion surgery originally designed to increase ICV are equally effective in reducing raised ICP with a more common cause—venous hypertension (see below).
Airway Obstruction
Impairment of the upper airway is common in the severely affected child with syndromic synostosis and is an important contributor to the vicious cycle that determines ICP in these children (see Fig 20.9).117 The peaks of ICP that can reduce cerebral perfusion pressure to as low as 14 mm Hg are invariably associated with episodes of airway obstruction during REM sleep. The practical importance of this is that improvement in the airway (e.g., by adenotonsillectomy,118 a nasopharyngeal prong, or tracheostomy) may help control ICP.
Hydrocephalus and Chiari 1 Deformity
As many as 40% of children with syndromic synostosis have a degree of ventricular enlargement,104 but in many it is nonprogressive. It is important, therefore, that the craniofacial surgeon does not proceed to treatment—a shunt insertion, for example—unless ventriculomegaly is progressive and other indicators of raised ICP are present.
Hydrocephalus occurs particularly when there is constriction of the skull base and early closure of the lambdoid sutures, which explains why it occurs more frequently in Crouzon and Pfeiffer (types 2 and 3) syndromes than in Apert syndrome.119
Although the fourth ventricle is usually small compared with the third, our experience (and that of others120) of endoscopic third ventriculostomy has not been encouraging, and we prefer to insert a ventriculoperitoneal shunt, particularly in a young child with very high pressure.
Constriction of the skull base, hydrocephalus and raised ICP, and herniation of the cerebellar tonsils (the Chiari 1 deformity) are linked in a cycle of cause and effect.119,121 The Chiari 1 deformity is seen most often in children with a constricted skull base, and although its progression (with additional buckling of the lower brainstem) is facilitated by raised ICP, it is also a risk factor for the development of hydrocephalus.122
When tonsillar herniation and brainstem distortion in a child with syndromic synostosis become symptomatic (usually when respiratory studies suggest a central as well as an obstructive contribution to breathing problems123; see below), a foramen magnum decompression may be needed.
Raised Venous Pressure
Intracanial venous hypertension is a major contributor to raised ICP in children with syndromic synostosis.124 Its cause is narrowing or actual occlusion of venous channels through the skull base125 that impedes the outflow of blood from the cranial compartment. The rise in venous pressure thus produced is aggravated by airway obstruction, and by interfering with CSF absorption, it is also a potential contributor to hydrocephalus—a situation similar to that often seen in achondroplasia.126
In a digital subtraction angiography study of 23 of our children with syndromic synostosis and raised ICP,127 18 had either complete or more than 50% occlusion of the sigmoid/jugular complex on one or both sides (▶ Fig. 20.12). Extensive collaterals through the retromastoid region and other transosseous channels can cause troublesome bleeding during reflection of the extensive skin flaps often required for craniofacial surgery.44,128
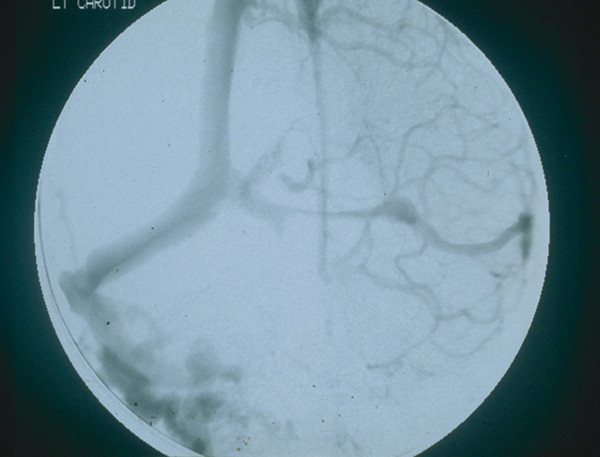
Fig. 20.12 Digital subtraction angiogram of a child with syndromic craniosynostosis showing near-occlusion of the left transverse sinus, absence of both sigmoid/jugular complexes, and a mesh of collateral veins palpable in the right retromastoid region.
Treatment would ideally either open up or bypass the obstructed venous channels. Although this has not proved successful,129 any vault-expanding procedure will lower ICP due to venous hypertension—another example of the close relationship among the various causes of raised ICP in syndromic synostosis.
In summary, raised ICP does not develop in all children with syndromic synostosis (about 40% of our children with Crouzon syndrome and only one-third of those in Renier’s series had an ICP above 15 mm Hg95), and not all who do are best treated by cranial vault expansion. This raises questions about the policy of many craniofacial units—the routine use of the fronto-orbital advance when these children first present.
20.4.2 Airway Obstruction
Impaired respiratory function, particularly at night, when snoring is a frequent complaint, is a common problem for the more severely affected child with syndromic craniosynostosis. It is usually caused by airway obstruction due to narrowed nares, cramped nasal passages, a maxilla constricted in all planes, and tracheal softening; however, a central component occurs when there is brainstem compression from a Chiari 1 deformity.123 An airway clear at birth may become obstructed during later growth of tonsillar and (in particular) adenoidal tissue in the restricted space available.
In addition to contributing to raised ICP, breathing difficulties impair the ability of the infant and young child to feed and are an important contributor to their failure to thrive. In older children, disturbed nights lead to sleepiness during the day and can interfere with schooling.
All children in whom breathing difficulties are suspected should undergo an overnight respiratory sleep study. Although the most common cause is upper airway obstruction, such a study will determine whether a central component is present; if there is a central component and it is associated with a Chiari 1 deformity, a foramen magnum decompression may be required.123
Management involves (in ascending order of magnitude) the following: insertion of a nasopharyngeal airway,130 adenotonsillectomy (which may need to be repeated as the child grows),118 continuous positive airway pressure (CPAP),131,132 tracheostomy, and finally operations that open the airway by advancing the maxilla—the Le Fort III advance (performed at an average age of 8 years in Fearon’s series133) and the monobloc frontofacial advance134 with or without distraction—and widening it (the facial bipartition).
20.4.3 Corneal Exposure
Recession of the maxilla below and the fronto-orbital region above can leave the corneas exposed and in danger of permanent scarring.
Temporary measures to protect them include the instillation of lubricating drops (particularly useful when the eyes do not close completely at night) and tarsorrhaphy, although this can raise the intraocular pressure in the face of severe exorbitism. Longer-term protection requires advancement of the bony orbital rim, either in part with a fronto-orbital advance or completely with a fronto-orbital advance combined with a Le Fort III maxillary advance or a frontofacial monobloc procedure (which can in exceptional circumstances be performed as a semi-emergency procedure135).
20.4.4 Cosmesis
The cosmetic disabilities that most trouble patients with syndromic synostosis and their families include a misshapen forehead, protruding eyes, eyes set too far apart (hypertelorism), and an upper jaw that is set back while the lower jaw protrudes.
In correcting for cosmesis alone, it is important to bear in mind the caveat already presented—that surgery carried out on a part of the craniofacial skeleton that is still growing may need to be repeated either wholly or in part in order to achieve a result that will prove stable over time. Each of the various components of the craniofacial skeleton has its own growth pattern. Waitzman et al have calculated that the cranio-orbito-zygomatic skeleton reaches more than 85% of its adult size by the age of 5 years.136,137 Our own policy, based more on clinical observation than on measurement, is to assume that a forehead and supraorbital region in a satisfactory configuration at around 10 years of age are unlikely to need further correction, and that cosmetic reconstructions after that age can focus more on the maxilla and mandible, where growth will continue until secondary dentition is complete during the mid to late teens.
20.5 Craniofacial Operations for the Child with Syndromic Craniosynostosis
Paul Tessier, who was a plastic surgeon, was the first to combine the skills of the plastic surgeon and those of the neurosurgeon to create what we recognize today as the specialty of craniofacial surgery.138,139 Indeed, all the procedures to be described here, if not actually designed by him, were introduced with his approval and incorporated into his extensive surgical repertoire.
20.5.1 General Points
Although strictly outside the scope of this chapter, the following points should always be considered before a child with a craniosynostosis-associated syndrome is taken to surgery.
Anesthesia
Craniofacial surgery should never be undertaken without the input of pediatric anesthesiologists experienced in the pre-, intra- and postoperative care of children who may already have an obstructed airway and raised ICP, and who (because of anomalous cranial venous drainage140) may be at risk for major hemorrhage during the procedure.141
An example of their preoperative role is providing input regarding the decision of whether a child with an obstructed airway who is about to undergo major frontofacial surgery should first have a tracheostomy.
General anesthesia for a patient with a severe facial deformity may also call for fiberoptic intubation, an essential skill for the craniofacial anesthesiologist.
The circulating blood volume of a young child is small, and losses that an older patient can withstand can lead to a swift circulatory collapse. A fail-safe approach is always to be ahead rather than behind concerning blood loss. Large-volume transfusions (one volume of circulating blood or more) will interfere with coagulation, and immediate access to fresh frozen plasma, platelets, and other clotting factors is essential.
Other ways of reducing the need for transfusion—or for the number of donors to whom the child is exposed—include the preoperative use of iron supplements (to ensure the child comes to surgery with an optimum haemaglobin level), the (still controversial) prescription of EPO (erythropoetin), and (during surgery itself) the use of cell-savers and antifibrinolytics.
Correct positioning of the patient is vital in craniofacial operations, and the anesthesiologist plays an important role in this. A head-up tilt with no obstruction of the neck veins is essential to reduce intracranial venous hypertension, although such a position will expose the patient to a (in our experience) small risk for air embolism. When surgery is performed with the patient prone, great care must be taken to protect the often protruding eyes and avoid any abdominal compression that may impede venous return.
Incision
Access
Access to large areas of the skull and face is often required in craniofacial surgery. We favor a bicoronal incision whose center lies just behind the hairline and then falls in a gentle curve to behind each ear. This provides access to the entire skull and, thanks to the pliant skin of the young, also to the upper reaches of the face. The hair need only be braided and can be left unshaved.142 Unless dissection to the zygomatic arch through the layers of temporalis fascia is required, the temporalis muscle is elevated with the skin flap. There is then no need to resuspend it at the end of surgery, and the frontal branch of the facial nerve is protected.
Cosmesis
A scar on the head is best hidden if the hair lies across it rather than being parted by it—as happens with the “classic” neurosurgical bicoronal incision, which follows the line of the coronal sutures downward to the front of the ear. Many units employ a zigzag incision to provide both hair cover and the degree of skin stretch often needed when the skull vault has been expanded.
Infiltration
For many years, we have injected a large volume of a “tumescent” solution containing two types of local anesthetic, a steroid, adrenalin, and hyalase into the proposed incision, the skin flaps, and when indicated, the tissues of the face. We have found that this reduces postoperative swelling and also shortened hospital stay.143
Complications
Of particular importance to the craniofacial surgeon are the following.
Cerebrospinal Fluid Leaks
Dural tears are likely to occur during craniofacial operations when there has been previous surgery (particularly if metallic plates and screws have had time to migrate inward144) and when the skull base is very constricted. Osteotomy cuts through the anterior skull base place the frontal extradural compartment in communication with the (bacterially contaminated) nose, allowing CSF leakage that presents as rhinorrhea. The patient is then at risk for meningitis, and contamination of the extradural space can lead to infection of the often devascularized surrounding bone.
Measures to reduce the risk for CSF leakage complicating monobloc and similar procedures include (in addition to previous experience145) the following: prophylactic placement of a lumbar drain at the start or end of the operation, placement of the skull base osteotomy no farther posteriorly than the foramen cecum, careful attention to the closure of any dural tears, covering the gap in the anterior skull base before the frontal bone is replaced with a vascularized pericranial flap, and finally the use of tissue adhesive to seal the area.
Fortunately, most CSF leaks cease spontaneously. Those that show no sign of settling over a day or so should be treated by the insertion of a lumbar drain. Leaks that persist (or recur) despite this may require a formal skull base repair, either transcranially or transnasally.
Infection and the Dead Space
The monobloc procedure by definition places the frontal extradural space and the nasal cavity in communication, something that a separation of the surgical components into a fronto-orbital advance and a Le Fort III advance does not do. However, any operation that increases the ICV carries the risk of leaving an air, blood, and serous fluid–filled dead space,146 which together with the often devascularized bone surrounding it provides an excellent substrate for bacterial growth.
Discussion with the hospital’s microbiologists is obligatory, both to determine the most suitable antibiotic regime to be used both prophylactically and to decide when infection is either confirmed or assumed in the absence of an obvious source.
In our unit, we have a low threshold for reopening the skin incision and thoroughly washing the surgical field with an antibacterial solution if infection is suspected.
Bone Defects
Cranial vault expansion often leaves areas of bone defect that in a child older than 1 or 2147 years of age are unlikely to fill in spontaneously. We regularly use a “salami” of milled bone fragments that are mixed with tissue adhesive and then “rolled out” in a thin strip to provide permanent bone cover for such defects.148
For the elective closure of bone defects in the older child we use when possible split calvarial bone.149
Bone Fixation
The tendency of metallic plates and screws to migrate inward144 has led us to avoid their use when possible, particularly in young children in whom sufficiently rigid fixation can usually be obtained with absorbable sutures. Metal plates and screws complicate subsequent operations when they become buried in bone; they sometimes eventually penetrate the dura and make tears that are difficult to repair inevitable during subsequent operations.
20.5.2 Craniofacial Procedures
The operations involved in the management of children with craniosynostosis syndromes cross many surgical specialties. Described here are those that include a neurosurgical element wholly or in part.
Fronto-orbital Advance
The operation most frequently performed in the management of syndromic synostosis is the fronto-orbital advance, in which the frontal bone and the supraorbital ridge are brought forward a couple of centimeters or so and then fixed in their new position. The fronto-orbital advance both expands the intracranial volume and provides cover for the upper parts of the globes. Its cosmetic effect may be all that is required in the management of less severely affected children—some with Muenke or Saethre-Chotzen syndrome, for example. At one time, it was believed that the skull base osteotomies involved would “free” the facial skeleton from the skull base, allowing the maxilla to assume a more normal trajectory—the so-called “floating forehead.”150 However, as discussed earlier, it is now recognized that maxillary (and fronto-orbital) growth is determined more by the patient’s genotype than by the effects of any frontal and base-of-skull disconnection.39,86
When both coronal synostosis and maxillary hypoplasia require attention (as in the Crouzon, Apert, and Pfeiffer syndromes), the fronto-orbital advance can be combined with a Le Fort III89 to advance the whole frontomaxillary complex as an alternative to the monobloc procedure.
Cranial Vault Expansion
The cranial vault is expanded anteriorly as a result of both the fronto-orbital advance (see above) and the monobloc procedure. However, biparietal and posterior vault expansions are also useful ways of lowering ICP due to craniocerebral disproportion with or without venous hypertension. A posterior expansion can when necessary be taken as far down as the foramen magnum to decompress a symptomatic Chiari 1 malformation. The released parietal bones or posterior vault can be left to float freely, but a greater increase in ICV is achieved if the bones are secured in their expanded position with bone struts and absorbable sutures. The degree of expansion achievable is limited by how far the overlying skin will stretch to accommodate the expansion, although such maneuvers as “relaxing” incisions in the galea and the use of looped tension sutures can ease edge approximation. The use of springs and distraction, however (see below), addresses this issue.
A cranial vault expansion may have unintended consequences, not the least of which is the conversion of a previously large but stable ventricular size into overt hydrocephalus.151
Springs Technology
The use of springs to move mobilized segments of the skull into a new position was introduced into craniofacial practice by Lauritzen et al.152,153
We have also found springs a convenient way of providing a greater degree of vault expansion because of their gradual as opposed to immediate intraoperative stretching of the overlying skin (▶ Fig. 20.13). The scale of the surgery required to insert them is often less than that of a conventional vault expansion, but another operation to remove them once bone consolidation has been achieved is usually required.
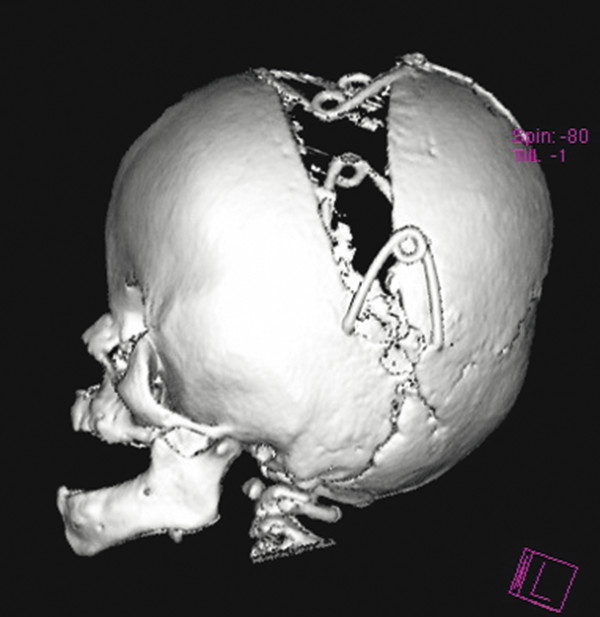
Fig. 20.13 A posterior vault expansion in a child with Apert syndrome performed with our unit’s springs technology.
(Image provided courtesy of Mr. Owase Jeelani, FRCS.)




