Fig. 1
Overlapping AAV genome design and transduction. (a) The transgenic cassette, containing the depicted elements, is split into two separate vectors such that there is a homologous sequence overlap (gray ). The total size of the transgenic DNA must be less than 4.7 kb. (b) Following co-transduction of vector genomes A and B, homologous recombination at the overlapping sequence occurs to reconstruct the depicted large transgenic DNA cassette of approximately 9 kb. Black box AAV inverted terminal repeat
1.4 Trans-splicing Technique
The trans-splicing technique also relies on a co-transduction approach in which the transgene is split such that one vector (vector A) contains a promoter, a 5′ portion of the gene, followed by a splice donor sequence (Fig. 2). The downstream vector (vector B) contains a splice acceptor sequence the remaining 3′ cDNA (or gene), and a poly A tail [4]. A concatemerization event in the correct orientation (theoretically about a 16 % chance) generates a single DNA molecule containing the intended large transgene expression cassette (Fig. 2) [4, 24]. Upon production of the pre-mRNA the intron is spliced out along with the ITR recombination junction, establishing an intact open reading frame large than can be packaged in a single vector [23, 25]. The trans-splicing method has been used in several Duchenne muscular dystrophy mouse models and has shown detectable restoration levels of dystrophin post-transduction [26–28]. There are also studies showing efficient whole-body and retina transduction that may also be promising for efficiently restoring large transgene expression [7, 29]. Even though efficient transduction is noticed in these experiments the transduction efficiency of these trans-splicing vectors is decreased compared to that of a single intact vector in skeletal muscle, eye, and liver [9]. The reason for this decreased transduction efficiency is due to several factors including (1) the efficiency of multiple vector transduction of a single cell, (2) the efficiency of the cellular machinery to reconstruct the large transgene in the desired orientation, (3) the decreased accumulation of mRNA due to the instability of the pre-mRNA, and (4) the gene splitting site [30]. The use of synthetic introns, including exonic splicing enhancers, can be used to at least partially counteract these rate-limiting steps [26, 31, 32].
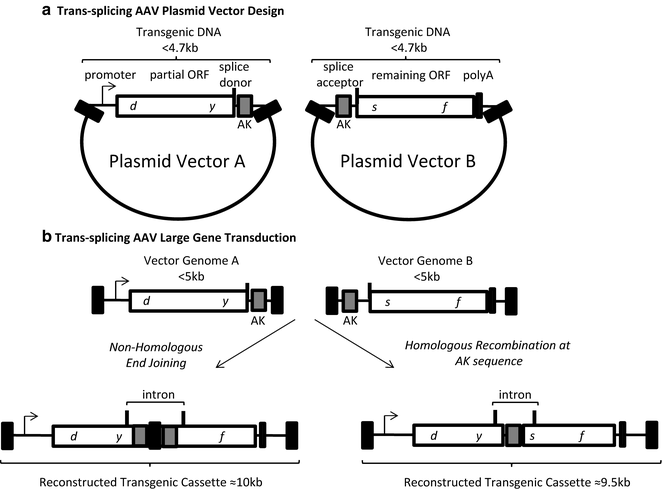
Fig. 2
Trans-splicing AAV vector design and transduction. (a) The genetic elements of the two required AAV plasmids are depicted. Plasmid A contains a promoter, a partial open reading frame terminated at a splice donor site, followed by a recombinogenic sequence (hybrid vectors only, AK). The vector B plasmid contains a recombinogenic sequence (hybrid vectors only, AK), a splice acceptor site, the remaining ORF followed by a poly A sequence. The transgenic DNA in each plasmid is less than 4 kb. (b) Following co-transduction vector genomes A and B can undergo end joining (left ) or homologous recombination at the AK sequence (right ) to reconstruct a large transgenic cassette containing the depicted intron. Black box AAV inverted terminal repeat
1.5 Hybrid Trans-splicing Technique
The hybrid trans-splicing technique is essentially a combination of the overlapping and trans-splicing large gene delivery approaches described above. These hybrid vectors are designed in the same manner as trans-splicing vectors with one key difference: the inclusion of an overlapping sequence within the intron of both the 5′ and 3′ vectors (Fig. 2) [6, 13]. To increase the likelihood of the desired large gene reconstruction, particular sequences that are considered “recombinogenic” can be used as the overlapping sequence [6, 12, 13, 33]. Following co-transduction, the full-length transgene can either be formed by HR at the overlapping sequence or the ITR homology. Alternatively, the ITRs may join via NHEJ [9, 20]. Then, transcription, splicing and translation generate the desired protein as described above for trans-splicing vectors. Current reports comparing different overlapping “recombinogenic” sequences within the hybrid trans-splicing strategy suggest that a 77 nt sequence derived from the F1 phage genome is the most efficient in promoting large transgene reconstruction, at least in the tested cells [12]. This hybrid AK vector context, as well as the normal trans-splicing approach, has demonstrated relevance for the treatment of retinal diseases including Stargardt’s disease and Usher syndrome type 1B [12].
1.6 Fragment AAV Large Gene Delivery
Regarding rAAV large gene delivery , reports have demonstrated the ability of the AAV capsid to package genomes over 5 kb [34–37]. For instance, Allocca et al. surveyed the packaging capacity of different capsid serotypes [34]. In the eye, it was demonstrated that AAV serotype 5 capsid, in particular, could package an expression cassette containing the adenosine triphosphate-binding cassette (ABCA4) at an unprecedented size of 8.9 kb, which was verified by alkaline gel electrophoresis [34]. Importantly, this AAV5-ABCA4 vector mediated effective transduction and resulted in the correction of disease phenotypes observed in the retina of a Stargardt’s disease mouse model [34]. The excitement for this groundbreaking report was later tempered by the reports of several groups that concluded that rAAV genomes over 5 kb were not packaged in their entirety, but instead DNA “fragments” of the intended oversized cassette were encapsidated (Fig. 3). Characterization of this process, demonstrated that oversized genome packaging initiates from the 3′ end and the external 5′ end is truncated when the capacity of the capsid is reached [8, 10, 11]. However, this process remains largely uncharacterized as heterogeneous DNA species (all <5 kb) are packaged in the apparently intact AAV capsids [8–11, 38]. Interestingly, these fAAV preparations retained their ability to mediate large gene transduction, albeit at a reduced efficiency compared to intact AAV in a tissue- and administration-dependent manner. A mechanistic evaluation of fAAV transduction in cell culture suggested that a large gene reconstruction event relies on HR, as the repair proteins Rad51C and XRCC3 were necessary while the NHEJ mediator DNA-PKcs was dispensable [9]. Despite the lack of an explanation for the earlier report demonstrating AAV5 large gene packaging [34], what is clearly evident is that a new rAAV large gene strategy emerged (fAAV) which, as in the other strategies, relies on the intracellular reconstruction of the oversized genome fragments, perhaps by canonical homology directed repair, at least in vitro [1, 9].
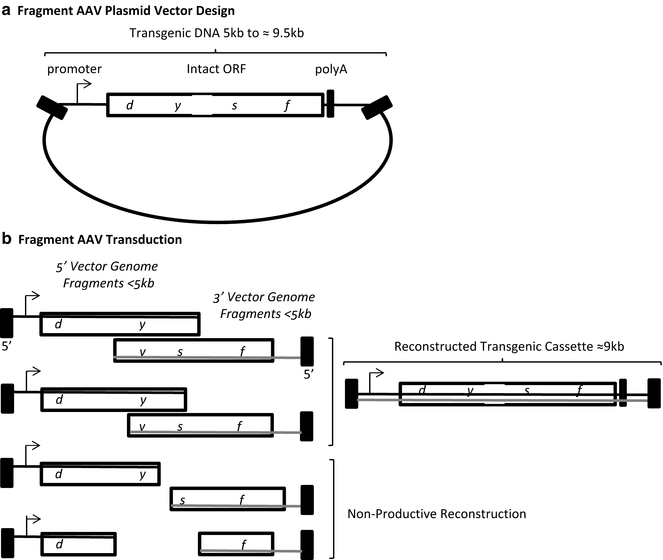
Fig. 3
Fragment AAV vector design and transduction. (a) A single plasmid with an intact transgenic cassette is depicted. (b) During AAV vector production different sized genome fragments of the over-sized transgenic cassette are packaged. Fragments of both polarities are packaged (gray and black lines) starting at the 3′ end. Opposite polarity fragments containing regions of complementarity can anneal followed by strand synthesis to generate a large transgenic cassette. Contaminating fragment genome species below half of the intended transgenic cassette do not contain strand complementarity and are non-productive for transduction. Black box AAV inverted terminal repeat
2 Materials
2.1 rAAV Production
1.
2× HEPES-buffer saline: 0.28 M NaCl, 0.05 M HEPES, 1.5 mM Na2HpO4, titrate to pH 7.05 with 5 N NaOH.
2.
Polyethylenimine (PEI) linear MW 25,000: 1 mg of PEI in 1 ml of 1× PBS, Adjust pH to 4 or 5 with 12 N HCl (see Note 1 ).
3.
DMEM: 10 % FBS, 1× penicillin/streptomycin.
4.
DNaseI digestion buffer: 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 50 U/ml DNase I.
5.
Sonicator (Branson Sonifier 250, or equivalent).
6.
Beckman Quick-Seal polypropylene (16 mm × 76 mm) centrifuge tubes (Beckman, Indianapolis IN, USA).
7.
Ultracentrifuge (Sorvall Discovery 90SE centrifuge, or equivalent).
8.
Ultracentrifuge rotor (Beckman NVT65 rotor, or equivalent).
9.
15 cm tissue culture plates.
10.
1.7 ml polystyrene microfuge tubes.
11.
Slide-A-Lyzer dialysis cassette with 20,000 (MWCO) and 0.5–3.0 ml capacity.
12.
HEK 293 cells.
2.2 Southern Dot Blot
1.
DNaseI digestion buffer (6 ml): 5.808 ml ddH2O, 60 μl DNase (10 μg/μl), 60 μl 1 M Tris pH 7.5, 60 μl MgCl2, 12 μl 1 M CaCl2.
2.
Proteinase K solution (10 ml solution): 6.9 ml ddH2O, 2 ml 5 M NaCl, 1 ml 10 % Sarkosyl, 100 μl proteinase K (10μg/μl).
3.
0.5 M NaOH (10 ml): 1 ml 5 M NaOH, 9 ml ddH2O.
4.
0.4 M Tris pH 7.5 (100 ml): 40 ml 1 M Tris pH 7.5, 60 ml ddH2O.
5.
Hybond-XL nylon membrane (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
6.
Filter paper.
7.
96-well dot blot manifold (or equivalent).
8.
Church buffer.
10.
High-salt wash (500 ml solution): 445 ml ddH2O, 50 ml 20 % SSC, 5 ml 10 % SDS.
11.
Low-salt wash (500 ml system): 492.5 ml ddH2O, 2.5 ml 20× SSC, 5 ml 10 % SDS.
2.3 Alkaline Gel Southern Blot
1.
Loading dye (6×): 80 μl 5 M NaOH, 10 μl 0.5 M EDTA, 0.18 g Ficoll, 730 μl H2O, xylene cyanol for color.
2.
Alkaline gel: Add 0.5 g agarose to 50 ml water (1 %), microwave to dissolve, let cool then add 500 μl 5 M NaOH and 100 μl 0.5 M EDTA (see Note 3 ).
3.
Gel running buffer: 10 ml 5 M NaOH, 2 ml 0.5 M EDTA, 988 ml H2O.
4.
Transfer buffer: 80 ml of 5 M NaOH in 920 ml of deionized water.
5.
Hybond-XL nylon membrane the size of the gel.
6.
Two sheets of filter paper slightly bigger than the size of the gel.
7.
One sheet of filter paper the width of the gel and about 1½ ft long for the transfer bridge.
8.
A pan that can hold the transfer buffer.
9.
A stack of paper towels.
2.4 Quantitative Polymerase Chain Reaction
3 Methods
3.1 Split rAAV Vectors: Construct Design
1.
Overlapping technique: To design your two split overlapping vectors, first construct plasmid vector A by placing a promoter followed by the transgene such that the total transgenic sequence is <4.7 kb (excluding the 2 ITR sequences which are each about 150 nucleotides) (Fig. 1). Next, place the remaining transgene sequence on plasmid vector B followed by a poly-adenylation sequence (Fig. 1). Again, the total size of the transgenic DNA needs to be <4.7 kb. Realistically, the overall size of the transgene cassette will dictate the maximum region of vector genome overlap (which is a requirement), with the basic understanding that increased region of homology will be more likely to stimulate intermolecular recombination , and thus the reconstructed large transgene cassette [23].
2.
Trans-splicing technique: Divide the transgene approximately near the middle, ideally at a conserved splicing junction. Then, add a canonical intron donor sequence immediately following the vector A ORF (Fig. 2). Any sequence between this donor signal and the vector A 3′ ITR will be intronic (Fig. 2). To generate vector B, an intronic sequence is positioned downstream of the 5′ ITR and terminated by an intron acceptor sequence (Fig. 2). Then, the remainder of the ORF (prematurely truncated in vector A is positioned upstream of a poly-A sequence prior to the 3′ ITR of vector B (Fig. 2)). As with the overlapping AAV vectors, and the overall rationale of this methods chapter, the total size of intact vectors (including the ITRs) needs to be <5 kb. Designs of trans-splicing constructs in the literature provide examples of the genetic arrangements and intronic sequences used previously [4, 6, 7, 12, 13, 24, 27].
3.
Hybrid trans-splicing technique: For this large gene AAV vector format, follow the design of trans-splicing vectors with a single exception, place the same overlapping sequence within the intron on the 3′ end of vector A and the 5′ end of vector B as depicted in Fig. 2. One consideration is the maintenance of a valid intronic branch point to facilitate splicing. Currently, there are only a few reports on overlapping sequences employed in hybrid AAV vectors and evaluation of the sequences used therein should be further investigated [6, 12, 13, 27].
4.
Fragment AAV vectors: construct design: A key distinction between fAAV large gene transduction and the split AAV vectors described above is that for the later approaches two distinct plasmid vectors (A and B) need to synthesized and employed for independent vector preparations whereas, a single plasmid containing the ITR sequences is used for fAAV (Fig. 3). Since DNA strands of both polarity are truncated and packaged during the viral vector production, the single plasmid design format is the following (5′ to 3′): (1) ITR, (2) promoter, (3) the oversized transgene, (4) a poly-A sequence, and (5) the flanking ITR sequence (Fig. 3). As genomic fragments will be packaged in both strand orientations, and need to anneal at a region of sequence complementarity, the total fAAV plasmid size between the ITR sequences should not exceed 9 kb.
3.2 rAAV Production
1.
24 h prior to transfection , split a confluent 15 cm plate of 293 cells at a 1:3 ratio into 4–6, 15 cm plates [final volume of culture medium (DMEM 10 % FBS 1× Penn/Strep) is approximately 20 ml].
2.
At approximately 70 % confluency, perform the triple transfection using polyethylenimine (PEI) using the plasmids and amounts described in Table 1 below and elsewhere [39].
Table 1
Transfection mixture for rAAV production
Transfection reagents | Amount per 15 cm plate |
|---|---|
pXX680 (Ad helper functions) | 12 μg |
pRepCap (replication and capsid genes) | 10 μg |
pITR-transgene (packaged sequence) | 6 μg |
Serum-free DMEM | 500 ml |
PEI | 100 μl |
3.
Add the three plasmids to a tube followed by the serum free DMEM and then the PEI. Immediately vortex the mixture and let sit at RT for 5 min. Add the transfection mixture dropwise to a single 15 cm plate of HEK 293 cells and gently swirl the plate to achieve uniform distribution.
4.
48–72 h post-transfection harvest the transfected 293 cells for intracellular vector purification (see Note 5 ).
6.
Pellet cells by centrifuging at 2500 rpm for 5 min.
7.
Remove the supernatant, wash the cells with 10 ml of 1×PBS, and spin again.
8.
Resuspend the washed cell pellet in 10 ml of 1×PBS and keep on ice.
9.
Sonicate each sample on ice for 25 pulses with a Branson Sonifier set at cycle 50 and output 5 (or equivalent).
3.3 rAAV CsCl Gradient Purification
1.
Following sonication, add 100 μl of the DNaseI solution to the samples and incubate at 37 °C for 1 h.
2.
 Lentivirus Production and Purification
Small-Scale Recombinant Adeno-Associated Virus Purification
Tissue-Specific Promoters in the CNS
Design of shRNA and miRNA for Delivery to the CNS
Altering Entry Site Preference of Lentiviral Vectors into Neuronal Cells by Pseudotyping with Envelope Glycoproteins
Nonviral Gene Therapy of the Nervous System: Electroporation
Lentivirus Production and Purification
Small-Scale Recombinant Adeno-Associated Virus Purification
Tissue-Specific Promoters in the CNS
Design of shRNA and miRNA for Delivery to the CNS
Altering Entry Site Preference of Lentiviral Vectors into Neuronal Cells by Pseudotyping with Envelope Glycoproteins
Nonviral Gene Therapy of the Nervous System: Electroporation




Bring samples to a final volume of 11.7 ml using 1×PBS.
Related posts:
Lentivirus Production and Purification
Small-Scale Recombinant Adeno-Associated Virus Purification
Tissue-Specific Promoters in the CNS
Design of shRNA and miRNA for Delivery to the CNS
Altering Entry Site Preference of Lentiviral Vectors into Neuronal Cells by Pseudotyping with Envelope Glycoproteins
Nonviral Gene Therapy of the Nervous System: Electroporation
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
