Chapter 8 Diagnosis and Surgical Options for Craniosynostosis
• In craniosynostosis, skull growth is arrested in the direction perpendicular to the fused suture and expanded at the sites of unaffected sutures (Virchow’s law), leading to characteristic calvarial deformations. In addition, the skull base and calvarial development are interrelated and changes at one location may affect the growth parameters at the other location.
• Intracranial hypertension can accompany craniosynostosis and is a function of the number of affected sutures, ranging from approximately 14% for single-suture synostosis to roughly 47% in multisuture synostosis. Children suspected of having elevated intracranial pressure may present with irritability, feeding difficulties, failure to thrive, headache, developmental delays, visual changes, calvarial towering, supraorbital recession, or lack of circumferential skull growth. Computed tomography (CT) scan changes may include “beaten copper” appearance of the inner table of the skull and compression of the ventricles and cisterns. Hydrocephalus and Chiari malformation can be associated with children with syndromic craniosynostosis (e.g., Crouzon, Apert, Pfeiffer syndromes).
• An increasing number of growth factor receptors (FGFR, TGF-βR), growth factors (FGF2, TGF-β, BMP), as well as transcription factors (MSX-2 and Twist), have been implicated in the pathogenesis of craniosynostosis and this list will undoubtedly grow in the future.
• The optimal timing of craniosynostosis surgery remains controversial even today, although the majority of craniofacial surgeons operate when patients are between 3 and 12 months of age. Because the normal brain and skull grow most rapidly in the first 2 years of life, early surgery takes advantage of this rapid period of growth and facilitates cranial volume expansion.
• Posterior deformational plagiocephaly, secondary to a supine sleeping position, will generally resolve with positional changes, physiotherapy, or helmet therapy and is only rarely a surgical condition.
Craniosynostosis is defined as the premature closure of a cranial suture which causes abnormal calvarial growth. Skull growth is arrested in the direction perpendicular to the fused suture and expanded at the sites of unaffected sutures, leading to characteristic calvarial deformations (Virchow’s law).1 In addition to the morphological changes accompanying craniosynostosis, functional problems related to brain development and possible intracranial hypertension are major considerations. Although the likelihood of elevated intracranial pressure remains low for patients with single-suture craniosynostosis, children with multiple-suture involvement or delayed presentation of single-suture synostosis are at significantly higher risk.2–4 A broad range of surgical options exist in the armamentarium of contemporary craniofacial surgical reconstruction, all with the primary objective of releasing the affected suture to permit normalization of skull growth in the setting of accelerated cerebral growth. Over time, progressively earlier recognition of craniosynostosis and its subsequent treatment have led to improved surgical results with correspondingly decreased perioperative morbidity. With a greater understanding of technologies relying on dynamic cranial vault alteration, including endoscopic sutural release, spring-assisted cranioplasty, and distraction osteogenesis, new horizons will inevitably unfold.
History
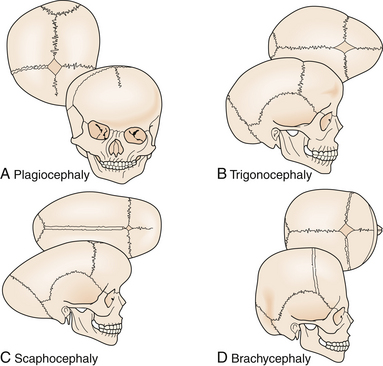
Craniosynostosis has long been recognized as an abnormal process originating at the calvarial suture. Early recognition of the importance of the skull sutures and their relationship to head shape was first made by investigators such as Hippocrates, Galen, and Celsus. In 1791, Sommerring noted that calvarial growth occurred at the suture line and that premature suture closure led to restriction of growth perpendicular to the affected suture.5 In addition to confirming Sommerring’s findings, Virchow was the first to describe the compensatory calvarial growth that occurred at the sites of unaffected sutures and associate a characteristic head shape with its corresponding abnormal suture (Fig. 8.1).1 These observations served as principal tenets directing craniosynostosis surgery over the subsequent century.
Over time, the relationship between calvarial growth and the skull base became better appreciated. In 1959, Moss pointed out the importance of the skull base in the promotion and development of the calvarial vault.6 His contributions included the observation that the cranial base developed prior to the calvarial vault and that characteristic abnormalities in the cranial base were associated with classic sutural abnormalities. Nevertheless, subsequent experimental work in animal models demonstrated that restriction of growth at specific sutures resulted in characteristic skull deformities that mimicked shapes seen in simple (nonsyndromic) craniosynostosis.7–9 As a result, the pathogenesis of craniosynostosis is currently thought to be a combination of skull base and calvarial growth disturbances.
Epidemiology
Craniosynostosis occurs in approximately 1 in 2000 to 1 in 2500 live births.10 This condition can be classified into simple (single-suture) versus complex (multiple sutures) or nonsyndromic versus syndromic (Table 8.1). Single-suture synostosis represents the majority of patients, with multiple-suture synostoses comprising approximately 5% to 15% of cases.10 As reported by large craniofacial centers, syndromic patients account for 15% to 20% of cases, whereas nonsyndromic patients constitute 80% to 85%.11
TABLE 8.1 Classification of Synostosis
| Affected Suture | Phenotypic Presentation |
|---|---|
| Sagittal | Dolichocephaly, scaphocephaly |
| Coronal (unilateral) | Anterior plagiocephaly |
| Coronal (bilateral) | Brachycephaly |
| Metopic | Trigonocephaly |
| Lambdoid | Posterior plagiocephaly |
| Multiple sutures | Cloverleaf (Kleeblatschädel), acrocephaly, oxycephaly |
Single-suture synostosis most frequently occurs sporadically, with familial aggregation accounting for 7% to 8% of sagittal and metopic synostosis.11 An equal frequency is found for all ethnic populations; however, gender predilection will vary depending on the type of suture pathology. The most commonly involved location is the sagittal suture, which accounts for 45% to 68% of all individuals12,13 and is marked by a male/female ratio ranging from 3.5:1 to 7:1.14 An autosomal dominant inheritance pattern with 38% penetrance was reported for sagittal synostosis.15 Metopic synostosis is now the second most common form of craniosynostosis (23.7-27.3% of cases), an observation that currently evades definitive etiopathogenesis, and shows a male predominance of 75%.11–13 Unicoronal synostosis, also known as anterior plagiocephaly, accounts for approximately 18% of patients with craniosynostosis,12 with girls outnumbering boys by a 3:2 ratio.16 Lambdoid suture synostosis, referred to as posterior plagiocephaly, is a relatively rare event in children with an observed incidence ranging from 0.9% to 4%.17–19 True lambdoid synostosis must be distinguished from posterior deformational plagiocephaly, also known as positional molding, in which there is occipital flattening on the affected side without associated suture fusion. This epiphenomenon is possibly related to the supine sleeping position in young children, instituted in 1992 to address sudden infant death syndrome (SIDS).20
Although the sporadic nature of simple craniosynostosis makes an accurate prediction of risk difficult to ascertain, it appears that the risk doubles for future siblings if there are no other family members involved. When one parent and child are affected, the subsequent risk rises to 50%. Conversely, if both parents are unaffected and two siblings are affected, the risk for additional sibling involvement approaches 25%.21
More than a hundred syndromes have been associated with craniosynostosis, often marked by an autosomal dominant mode of transmission.22 Among them, Crouzon, Apert, and Pfeiffer (Fig. 8.2) syndromes are the most frequently occurring. Syndromic synostosis is commonly associated with multiple suture closure (coronal, sagittal, etc.) combined with other systemic manifestations (Table 8.2).
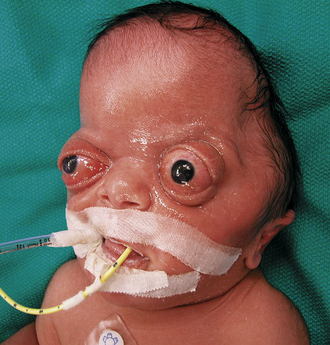
TABLE 8.2 Craniofacial Dysostosis Syndromes
| Syndrome | Involved Suture | Morphological Presentation |
|---|---|---|
| Crouzon | Coronal, sagittal | Midface hypoplasia, shallow orbits, proptosis, hypertelorism |
| Apert | Coronal, sagittal, lambdoid, others | Midface hypoplasia, shallow orbits, proptosis, hypertelorism, symmetrical syndactyly of hands and feet, choanal atresia, ventriculomegaly, genitourinary/cardiovascular anomalies |
| Pfeiffer | Coronal, sagittal | Midface hypoplasia, proptosis, hypertelorism, broad great toe/thumb |
Genetic and Etiological Factors
The etiology of craniosynostosis remains elusive because of its heterogeneous nature. Nevertheless, numerous factors are now known to promote or have been implicated in the development of premature closure of the calvarial sutures. Multiple teratogens, genetic mutations, metabolic disorders, and blood dyscrasias have been associated with craniosynostosis (Table 8.3). Interestingly, maternal smoking has been associated with isolated craniosynostosis23 and advanced paternal age has been found to trend with a higher frequency of metopic synostosis.12
TABLE 8.3 Recognized Causes of Craniosynostosis
| Hematologic disorders | Thalassemias Sickle cell anemia Polycythemia vera |
| Teratogens | Valproic acid Retinoic acid Aminopterin Diphenylhydantoin |
| Genetic conditions | |
| Metabolic disorders | Rickets Hyperthyroidism |
| Mucopolysaccharidoses | Hurler syndrome Morquio syndrome Mucolipidosis III |
| β-Glucuronidase deficiency | |
| Malformations | Holoprosencephaly Encephalocele Microcephaly Hydrocephalus (shunted) |
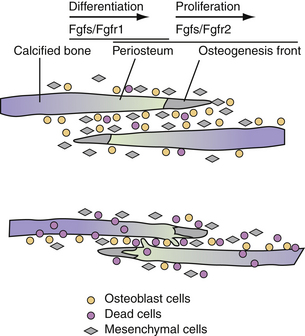
With advances in molecular genetics, candidate gene mutations as well as the molecular interactions underlying cranial deformities have been elucidated.24,25 Normal suture growth and morphogenesis is dependent upon a delicate balance between the proliferation of osteoprogenitors within the suture mesenchyme and differentiation to osteoblasts at the osteogenic fronts (Fig. 8.3).26 It is now known that the majority of syndromic craniosynostoses are caused by mutations in genes encoding fibroblast growth factor receptors (FGFR-1, FGFR-2, and FGFR-3) and the transcription factors Twist and MSX-2.27–33 Moreover, these same genes are responsible for approximately 25% of all cases of craniosynostosis.34 In general, gain-of-function mutations are associated with the MSX2 and FGFR genes, while loss of function or haplo-insufficiency abnormalities are found in TWIST gene mutations.35
Genetic alterations in FGFR-1 and FGFR-2 have been implicated in Crouzon, Apert, Pfeiffer, and Jackson-Weiss syndromes.30,36–39 FGFR-1 has been found to regulate osteoblast differentiation; therefore, a gain-of-function mutation may precipitate premature suture fusion through promotion of osteoblast differentiation and bone formation.40 Moreover, FGFR-2 has been linked to activation of osteogenic cell apoptosis. As shown by Chen and co-workers, a gain-in-function mutation leads to increased apoptosis and results in decreased cell numbers and distance between two overlapping bones. Ultimately, this develops into physical contact of two opposing bones, eventually leading to premature closure.41
Alterations in growth factor receptors have also been observed in nonsyndromic craniosynostoses. FGFR-3-associated coronal synostosis, also known as Muenke-type craniosynostosis, has been identified in up to 52% of patients with nonsyndromic bicoronal synostosis,42 either as a result of de novo mutations or associated with an autosomal dominant inheritance pattern.43 Gripp and colleagues observed that 10.8% of patients with unilateral coronal synostosis were positive for an FGFR-3 mutation and subsequently recommended testing of all patients with unilateral coronal synostosis to assess the risk of recurrence.44 These guidelines stem from the observation that Muenke-type craniosynostosis has been associated with a reoperation rate of at least 43%.45
Studies have implicated transforming growth factor-beta receptors (TGF-βR) in syndromic and intrauterine head constraint-related craniosynostosis. Loeys and co-workers have discovered a link between mutations of TGF-βR1 and TGF-βR2 and a syndrome of altered cardiovascular, craniofacial, neurocognitive, and skeletal development.46 Hunenko and colleagues demonstrated upregulation of TGF-βR1 and TGF-βR2 in mice undergoing intrauterine constraint leading to coronal suture synostosis.47 Such data points to the ability of mechanical forces to alter growth factor–mediated signaling during craniofacial growth and development.
In addition to the aforementioned growth factor receptors, their corresponding ligands have been found to be an integral component of calvarial osteoblast proliferation and subsequent sutural fusion. FGF2 has been shown to enhance proliferation rates in rat fetal osteoblasts, promote premature fusion of frontal sutures in calvarial organ cultures, and correlate with intrauterine constraint-related coronal suture synostosis.47,48 Inverse patterns of TGF-β isoform expression between fusing and patent sutures have been demonstrated in animal and human models. Opperman and colleagues demonstrated declining levels of TGF-β3 but continued expression of TGF-β1 and TGF-β2 posterior frontal suture fusion.49 Bone morphogenetic proteins (BMPs), members of the TGF-β superfamily, are involved in a broad range of developmental roles, including bone formation, skeletal patterning, and limb development. Several investigators have demonstrated the critical role of BMPs and their antagonists in dictating cranial suture biology. In particular, in situ hybridization of mouse cranial sutures localized expression of BMP-2 and BMP-4 to the osteogenic fronts and BMP-4 to suture mesenchyme and dura mater in the sagittal and posterior frontal sutures.50 Nacamuli and co-workers found BMP-3, a bone morphogenic protein antagonist, to be decreased in normally fusing posterior frontal sutures and increased in normally patent sagittal sutures.51
Mutations of transcription factors have also been implicated in causing syndromic forms of craniosynostosis. Liu and co-workers linked a gain-of-function mutation in the MSX-2 transcription factor with Boston-type craniosynostosis.52 Loss-of-function mutations in Twist proteins, transcription factors activating osteoblast differentiation, have been found to cause Saethre-Chotzen syndrome.28 Woods and colleagues have recommended TWIST1 mutation screening of all patients with either bicoronal or unicoronal synostosis, given that this genetic alteration confers a greater risk of recurrent intracranial hypertension and subsequent reoperation than nonsyndromic synostosis of the same sutures.53
Anatomical and Pathological Considerations
The calvarium grows most rapidly during the first 12 months, with the brain doubling in volume in the first 6 months and again by the second birthday. While calvarial expansion is most pronounced during the first 2 years, growth continues in a linear fashion until the age of 6 to 7 years, at which time the cranium is 90% of the adult size. Most of this cranial growth takes place in the sutures between the bone plates. Within the center of the sutural area, a population of proliferating osteoprogenitor cells is maintained. A portion of these cells enters the pathway of osteogenic differentiation, forming bone-matrix-secreting osteoblasts at the bone edges and contributing to skull expansion.54 Normal cranial suture closure occurs from front to back and from lateral to medial, with the metopic suture usually closing between 9 and 11 months of age55 and the remaining sutures fusing in adulthood.
A disturbance in the balance between proliferation, differentiation, and apoptosis causes premature ossification within the suture and its synostosis.56 Factors disturbing this balance include genetic or acquired changes in growth factor receptor/ligand profiles, loss of direct contact between dural and sutural cells, and increased external mechanical forces. As mentioned previously, many of the syndromic forms of craniosynostosis are attributed to alterations in the FGF/FGFR, TGF-β/TGF-βR, and BMP cascades. Both cerebral hypoplasia and overshunted hydrocephalus have been associated with secondary craniosynostosis, phenomena likely attributed to loss of dural contact.55,57 Both breach positioning and twin pregnancies have been associated with intrauterine constraint-related craniosynostosis, stemming from mechanical force signal transduction.58
Diagnostic Evaluation and Imaging
Preoperative assessment for craniosynostosis includes a detailed medical history, physical examination, and radiographic imaging. Medical history should elicit for asymmetrical calvarial deformities noted by friends or other family members, family history of calvarial deformities, and symptoms of intracranial hypertension (headache/vomiting, developmental changes, irritability, and oculomotor paresis). Physical examination should evaluate for characteristic calvarial shapes and asymmetries, premature closure of the anterior fontanelle (normally open until 12-18 months of age), perisutural ridging (calcification), and signs of intracranial hypertension (papilledema, supraorbital retrusion, severe towering, and severe frontal/occipital bossing). Routine funduscopic examination for papilledema is an accurate predictor of raised pressure in the older child but may not be 100% sensitive for the younger child (<8 years old).59 Craniofacial asymmetries should be documented in the form of head circumferences, cranial indices, and anthropometric measurements. The history combined with the examination is often confirmatory in an experienced primary care physician/nurse or craniofacial surgeon’s initial evaluation.
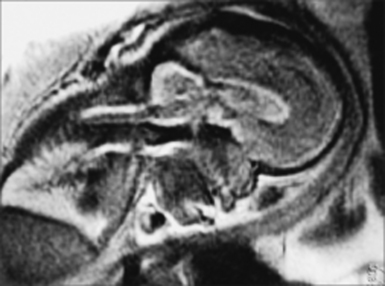
The role of radiological workup for craniosynostosis varies among clinicians. Currently, it is not uncommon for prenatal ultrasound to document craniosynostosis in utero.60–63 In addition to the ultrasound evaluation, fetal magnetic resonance imaging (MRI) (Fig. 8.4) at some centers has offered significant prenatal definition.64 Radiological investigation may be necessary to corroborate the diagnosis and rule out any associated intracranial abnormalities in the postnatal consultation period. Computed tomography (CT) studies remain the most sensitive barometer of bony fusion, as skull plain films suffer from poor sensitivity and a high false positive rate. The recent advent of three-dimensional CT (3D CT) has provided an excellent view of affected suture(s) as well as overall head shape, thereby simplifying the diagnosis and helping with surgical planning.60,65,66 This modality is not mandatory, but rather is reserved for multiple/complicated suture pathology, confirmation of diagnosis, or demonstration of skull base pathology.
CT scans may also provide radiological evidence for raised intracranial pressure. The presence of intracranial hypertension is dependent on the number of affected sutures, ranging from approximately 14% for single-suture synostosis to approximately 47% in multiple-suture synostosis, as well as on patient age.2,3 Although few children will manifest clinical symptoms of increased intracranial pressure, it is not uncommon to visualize erosion of the inner calvarial table (beaten copper appearance) on CT scan. A diffuse beaten copper appearance has been associated with greater intracranial pressure, as reported by Tuite and co-workers.67 Though it is common to see expanded subarachnoid spaces in all types of craniosynostoses, these spaces usually spontaneously resolve and are not felt to represent an increase in intracranial pressure.68 If there is any evidence for elevated pressure, surgical consideration should be expedited. This is especially vital in patients with syndromic synostosis (e.g., Crouzon, Apert, Pfeiffer syndrome), who are at a greater risk for hydrocephalus and Chiari malformations with associated intracranial hypertension.
CT and MRI studies are also helpful in evaluating the underlying brain for any structural or functional abnormalities. Unrecognized intracranial abnormalities may exist in a small number of patients and may include hydrocephalus (more common in patients with Crouzon syndrome), partial agenesis of the corpus callosum, holoprosencephaly (seen in patients with trigonocephaly), or focal cortical dysplasias. Indeed, as reported by Boop and colleagues, up to 5% of their patients with sagittal synostoses had unappreciated underlying intracranial pathology.69
Therapeutic Considerations
Surgical Indications
Correction of calvarial contour deformities and prevention of psychosocial dysfunction, intracranial hypertension, and mental retardation are the thrusts for surgical intervention in craniosynostosis. In the past, surgical intervention for simple craniosynostosis was undertaken primarily because of cosmetic and psychosocial considerations.70,71 Recently, sutural release in simple craniosynostosis has been advised owing to the concerns regarding raised intracranial pressure as well as mild but significant developmental delay in the aging child with uncorrected single-suture synostosis.2,3,72–74 In contrast to simple craniosynostosis, patients with complex or syndromic synostoses present with increased severity in neurological and cosmetic symptoms;3,75–77 therefore, surgical intervention in these infants is even more imperative.
Timing of Surgery
The optimal timing for reconstructive surgery in craniosynostosis remains controversial, as the age at surgery has different effects on intraoperative hemodynamics, postoperative cranial growth, and subsequent mental development. With regard to intraoperative hemodynamics, Meyer and co-workers demonstrated that older patient age (>6 months) was associated with decreased blood loss.78 In addition to benefiting from decreased blood loss, older infants can tolerate extensive blood loss better than younger infants. From the perspective of long-term skull growth, data are conflicting. In 1987, Whitaker demonstrated that as surgical age increased, the likelihood of secondary surgery also elevated.79 On the other hand, Fearon and colleagues recently found that older patient age (≥12 months) was associated with less diminished cranial growth following correction of all types of single sutural craniosynostosis.80 These findings must be weighed against the need to fully reconstruct any advanced postoperative skull defects in children over 12 months of age, because dura will not regenerate bone as readily. From the standpoint of mental development, Arnaud and co-workers reported that postoperative mental outcome was significantly better when surgery was performed before the patient reached 12 months of age.42
Although the literature is inconclusive regarding the appropriate timing for correction of craniosynostosis, the majority of craniofacial surgeons operate between 3 and 12 months of age. The specific time period is dependent on the type of surgical approach used. In general, endoscopic corrections are done at an earlier age, namely, by 3 to 4 months of age. Open surgical corrections are often done later. Fearon and colleagues perform treatment at 4 months of age for sagittal synostosis and 9 months of age for all other single-suture synostoses (metopic, coronal, and lambdoid).80 Marchac and associates reviewed their craniofacial experience with 983 patients operated on over 20 years, discussing their timing of surgical operations.81 Children with brachycephaly underwent a floating forehead procedure between 2 and 4 months of age, infants with sagittal synostosis underwent parasagittal craniectomies between 2 and 4 months of age or a frontocranial remodeling procedure if presenting between 6 and 9 months of age, and infants with either metopic or unicoronal synostoses had a frontocranial remodeling procedure between 6 and 9 months of age.
Type of Surgery
There is a growing debate in the literature regarding the optimal type of operation for correction of craniosynostosis. An open craniofacial approach was proposed as early as 1890, with Lannelogue advocating early open surgical release of a fused suture to prevent intracranial hypertension.82 Building on the principles of Lannelogue, a number of centers have reported their large-volume experience with open cranial vault remodeling procedures for all types of craniosynostoses.79,80,83–87 In 421 intracranial operations with movement of one or both orbits, Whitaker and co-workers in 1979 reported a 2.2% rate of mortality, 6.2% rate of infection, and 2.2% frequency of CSF leak.85 With increased experience and refinement in technique, Whitaker and colleagues reported a 0% mortality rate, 3.7% infection rate, and 1.2% CSF leak rate in a 1987 report of 164 open craniofacial procedures for craniosynostosis.79 Regarding longevity of the open craniofacial procedure, McCarthy and associates identified a 13.5% rate of reoperation for simple craniosynostoses and 36.8% revision rate for complex craniosynostoses during a 20-year experience.86,87 Reoperation rates have also decreased with experience and refinement of open surgical technique, with Sloan and co-workers reporting a 7.2% overall rate of reoperation in 250 patients and Fearon and associates noting a 2% rate of revision in 248 cases of simple craniosynostosis.80,84
Endoscopic Craniosynostosis Correction
Jimenez and Barone pioneered endoscopic sutural release in the mid-1990s,88–94 and it has been used by increasing numbers of surgeons who treat craniosynostosis. In general, the idea is to perform a minimally invasive strip craniectomy of the fused suture at an early age. The child then wears a cranial molding helmet, which slowly corrects the deformity over several months.
For coronal (Fig. 8.5) or metopic craniosynostosis, the child is positioned supine. One small incision is used to access the fused suture. A burr hole is drilled, and an endoscope is used to separate the dura from the overlying bone. Bone-cutting scissors are then used to cut out the fused suture. Irrigation and Gelfoam are used for hemostasis, and the incision is closed.
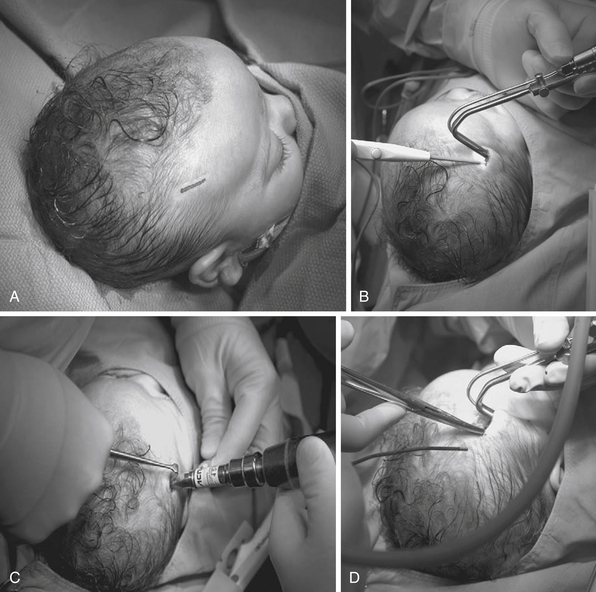
Postoperatively, patients are placed in orthotic helmets for 7 to 12 months to facilitate dynamic cranial vault alteration. Summarizing the data from studies by Jimenez and Barone,88–94 average age at operation is between 3 and 4 months, mean operating time centers around 60 minutes, average estimated blood loss is approximately 30 mL or approximately 5% estimated blood volume (EBV), transfusion rate hovers around 10%, average length of stay is generally 1 day, and complication rates range from 0% to 6%. These numbers are far lower than the blood loss of 25% to 500% of EBV, blood transfusion volume of 25% to 500% of EBV, and 4- to 7-day length of stay associated with open craniofacial techniques.93 MacKinnon and co-workers also found that children treated with endoscopic repair of unicoronal craniosynostosis may have less severe eye findings, such as V-pattern strabismus, than children treated with fronto-orbital advancement at a later age.95
Spring-Assisted Cranioplasty
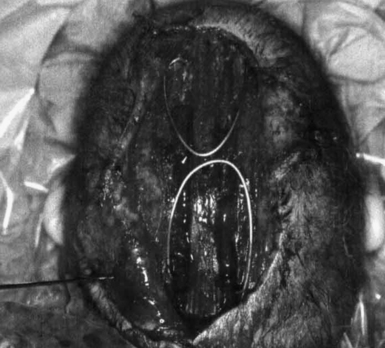
Spring-assisted cranioplasty was first undertaken on human subjects by Lauritzen in 1997, based upon success in the rabbit model by Persing and co-workers.96,97 This technique relies on a standard open access incision, osteotomies at the sites of stenosed sutures, placement of omega-shaped tension springs across the osteotomy sites, and possible placement of compressive springs along areas of compensatory growth (Fig. 8.6). Implantable springs are typically removed 4 to 7 months postoperatively. In 2008, Lauritzen and co-workers reported their large-volume experience with spring-assisted cranioplasty for all forms of craniofacial surgery.98 Results included an average operative time of 97 to 215 minutes, mean blood loss ranging from 143 to 503 mL, average length of stay between 5 and 6 days, mean cephalic index of 74 in scaphocephalic infants 6 months following cranioplasty, 6% reoperation rate, 3% rate of intracranial hypertension secondary to compressive springs, and a 0% mortality rate. On the heels of this study, David and co-workers reported their experience with the first 75 spring-assisted surgeries for scaphocephaly.99 With a mean follow-up period of 46 months, mean cephalic index was 75.4, which is comparable to patients with open cranial vault reconstruction and was maintained at 3- and 5-year follow-ups. Optimism with this emerging modality must be balanced against the need for a second operation for spring removal as well as the lack of control of spring action.
Distraction Osteogenesis
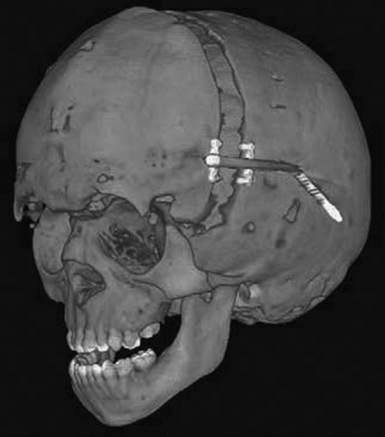
Along the lines of spring-assisted cranioplasty, distraction osteogenesis has been investigated for the treatment of craniosynostosis. This method entails a standard open access incision, osteotomies at the sites of stenosed sutures, and placement of internal versus external distraction devices (Fig. 8.7). After a latency period of 3 to 5 days, the devices are activated for a number of weeks until desired expansion and then followed by a 2- to 3-week consolidation period. Given that appraisal of this technique has been limited to the setting of small case reports,100–112 its safety and efficacy cannot be commented upon at this time.
Clinical Presentation/Therapeutic Considerations
Metopic Synostosis (Trigonocephaly)
Clinical Features
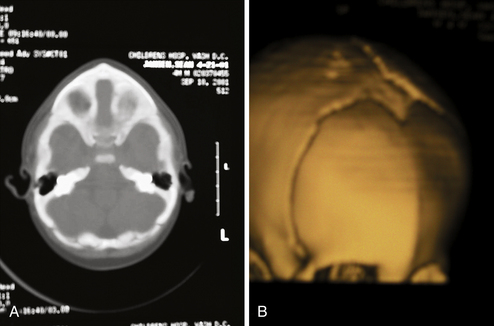
Metopic synostosis is often accompanied with a variable degree of phenotypic severity. Patients may present with mild ridging of the metopic suture, unaccompanied by other manifestations. Premature closure of the metopic suture may also lead to the formation of a triangular head, otherwise known as trigonocephaly. On the severe end of the spectrum, patients may present with a prominent “keel” forehead accompanied by recession of the lateral orbital rims, hypotelorism, and constriction of the anterior frontal fossa (Fig. 8.8).
Among the nonsyndromic craniosynostoses, metopic synostosis is most associated with chromosomal abnormalities, other brain malformations, and cognitive/behavioral dysfunction. Mild variants of metopic synostosis have been recently associated with abnormalities of chromosomes 3, 9, and 11.113–115 In addition, Tubbs and co-workers found a 30% incidence of type I Chiari malformations in the evaluation of patients with simple metopic ridges and postulated that these children were at greater risk secondary to the diminished anterior cranial volume.116 At the other end of the spectrum, severe cases of metopic synostosis have been associated with underlying frontal brain dysmorphology as well as other congenital anomalies.117
Metopic synostosis has also been associated with neurodevelopmental delay, with reported deficits ranging from modest to severe. Historically, metopic synostosis had been considered the form of single-suture synostosis with the highest degree of neuropsychological morbidity.118 Although Becker and co-workers confirmed a high prevalence of speech, cognitive, and behavioral abnormalities in patients with metopic synostosis, reported as 57%, they observed no differences in the degree of neuropsychological morbidity between different forms of single-suture synostoses.119 More recently, however, studies have concluded a modest to no delay in neurodevelopment.120,121 Da Costa and co-workers found that children with nonsyndromic craniosynostoses did not display obvious evidence of intellectual dysfunction, with mean intelligence quotients within the normal range. In 2007, Speltz and colleagues demonstrated a modest but reliable neurodevelopmental delay in children with all forms of single-suture synostoses in comparison to case-matched control subjects.121 Similar to Becker’s study, they found no difference in the extent of neuropsychological morbidity between different types of single-suture synostoses.119,121
Radiological Evaluation
Radiographic imaging by plain skull radiographs may demonstrate a hyperostotic, midline metopic suture in addition to hypoteloric orbits in severe examples of trigonocephaly. Nevertheless, definitive diagnosis is best made by CT, which will offer better bone definition while also evaluating the cerebral parenchyma (Fig. 8.9). Frontal dysmorphology is most commonly seen in this type of craniosynostosis and may consist of corpus callosum dysgenesis, holoprosencephaly, and other frontal dysembryogeneses. Patients suspected of harboring a Chiari malformation are best served by an MRI, although CT scans with low cuts through the posterior fossa may also demonstrate a crowded foramen magnum as a result of tonsillar herniation.
Surgical Therapeutics
Delashaw and colleagues proposed that metopic synostosis and trigonocephaly represent an embryological continuum, directing their surgical approach based on the severity of the frontal calvarial deformities.122 In general, the goals of surgery are the normalization of the forehead with reconstitution of a normal supraorbital rim if necessary.123–129 Individuals presenting solely with a prominent midline keel may be best served by simple contouring of the frontal bone or by removal of the frontal bone flap followed by reconfiguration. These children otherwise demonstrate normal orbital and supraorbital anatomy. Conversely, patients with significant trigonocephaly and hypotelorism will require a fronto-orbital reconstruction, recontouring the frontal bone and laterally expanding the orbits at the same time. Evolution of surgical technique has included more radical treatment of the involved sphenoid bone with simultaneous correction of the hypotelorism.130–132
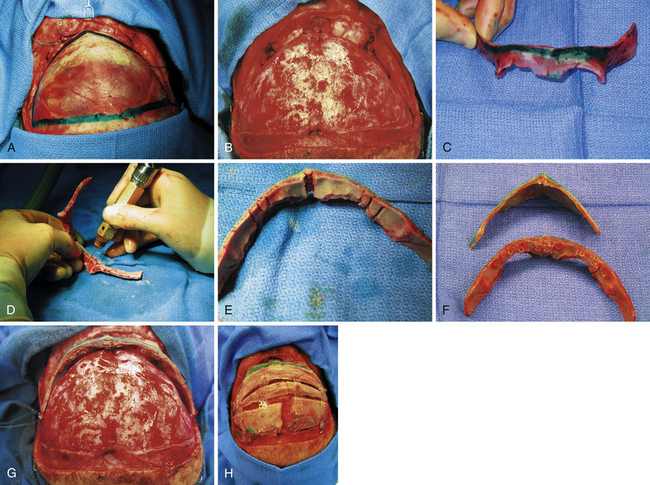
The essentials of fronto-orbital reconstruction involve a standard bicoronal incision, which will provide adequate exposure of the fronto-orbital region while minimizing any postoperative scar. Perioperative antibiotics (cefazolin 10-20 mg/kg loading dose, 8 mg/kg intravenously every 8 hours for 48 hours) as well as steroids (Decadron 0.25 mg/kg intravenous every 6 hours for 48 hours) are given. Prior to the start of surgery, bilateral tarsorrhaphies are undertaken. The incision is infiltrated with 0.5% lidocaine and 1:400,000 parts epinephrine to minimize intraoperative bleeding. The frontal and temporal regions are dissected in the subgaleal plane leaving the periosteum intact on the surface of the bone which also helps to minimize bleeding (Fig. 8.10A). The dissection is taken down to the level of the periorbital tissues, taking care to avoid any injury to the underlying globes. Following exposure of the frontal and orbital regions, the frontal bone is removed, providing access to the intracranial compartment (Fig. 8.10B). The supraorbital rim is then removed in one piece to facilitate reconstruction of the previously triangular supraorbital bar. Care is taken to remove sufficient bone in the region of the sphenoid bone to allow for growth at the midface and orbits (Fig. 8.10C). If the orbits require correction of hypotelorism, it will be necessary to displace the lateral walls of the orbit as well as to split the midline and interpose a calvarial bone graft. Reconfiguration of the supraorbital bar often requires a midline osteotomy to facilitate a flattened forehead with additional partial-thickness bone cuts at the lateral (pterional) angle to promote normalization of the lateral supraorbital angle (Fig. 8.10D and E). The supraorbital reconfiguration is maintained by the utilization of intervening bone grafts as well as absorbable hardware (Fig. 8.10F). Following placement of the supraorbital bar as a foundation (Fig. 8.10G), the frontal bone is reconstructed using the remaining portions of bone. It is often possible to reverse the original frontal bone flap (posterior portion now in an anterior position) to obtain an adequate width and contour with the new frontal bone flap. It is important to provide an adequate enhancement at the pterional region to avoid long-term supratemporal hollowing or recession. Reconstruction is facilitated with absorbable hardware, as permanent hardware has been associated with transcranial migration (Fig. 8.10H).
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


