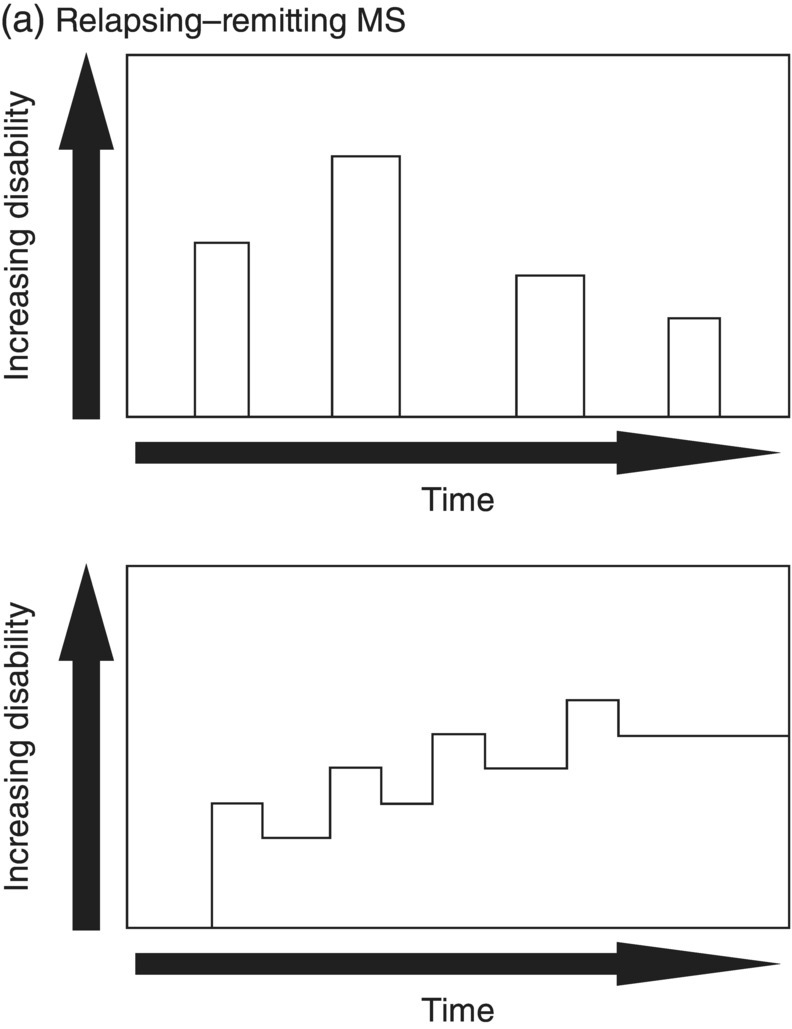
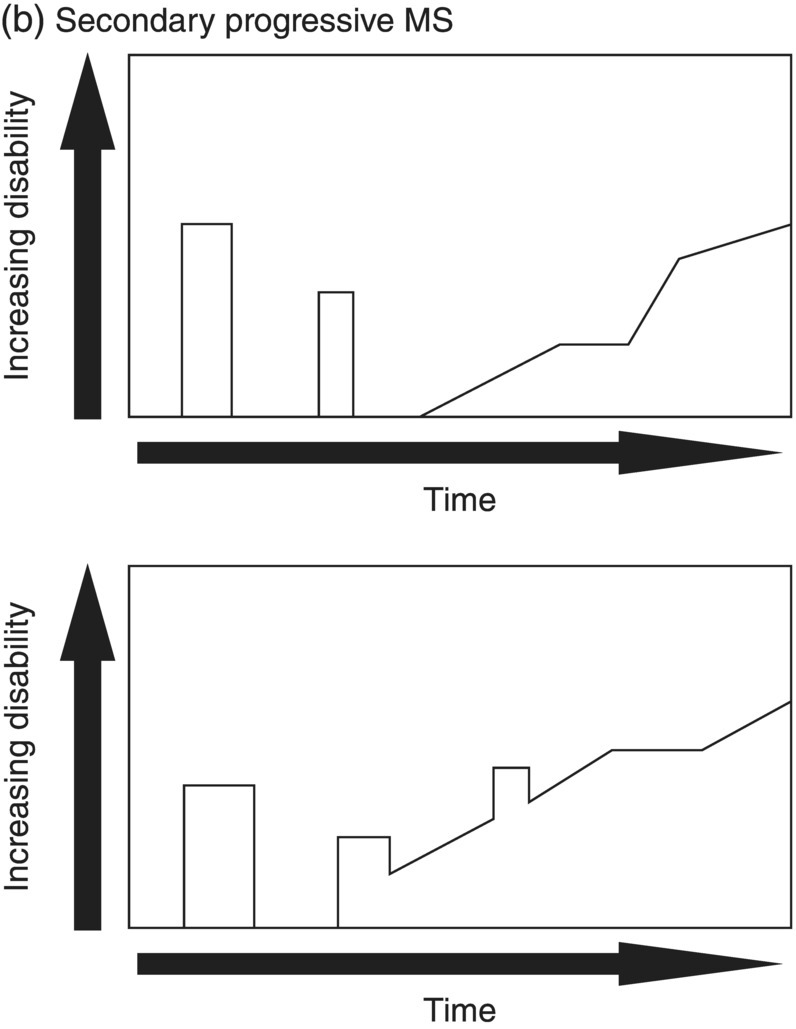
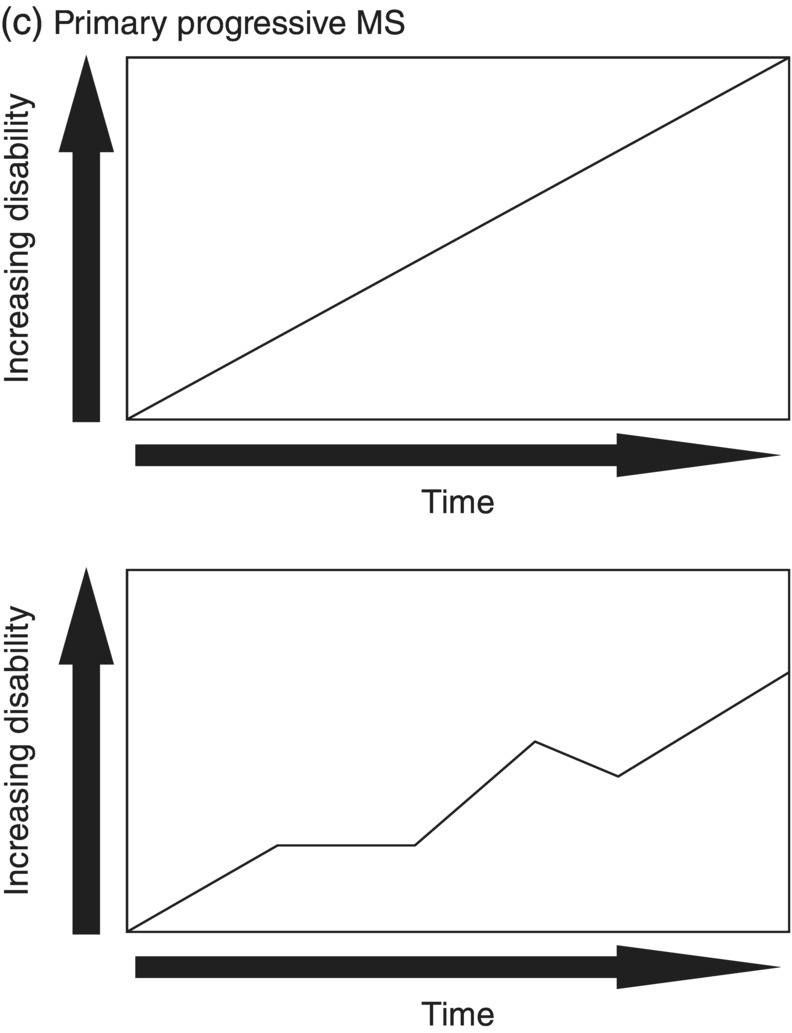
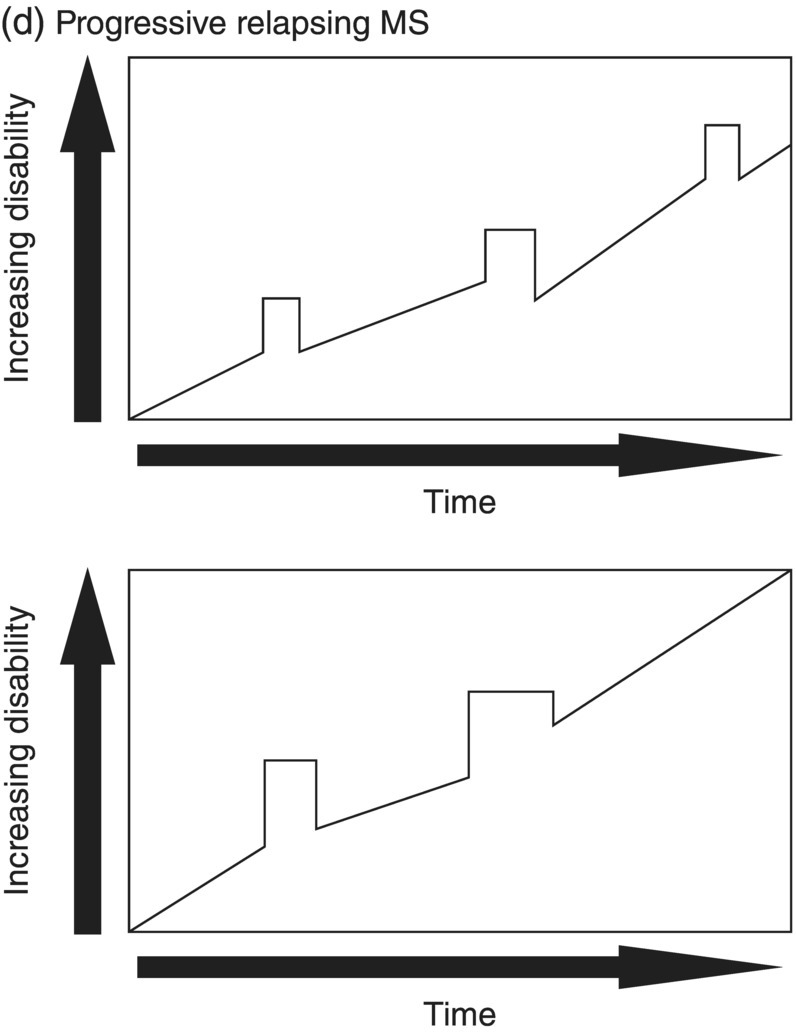
3 Dalia Rotstein1,2 and Paul O’Connor1,2 1 Division of Neurology, Institute of Medical Science, University of Toronto, Toronto, Ontario, Canada 2 St Michael’s Hospital, University of Toronto, Toronto, Ontario, Canada Multiple sclerosis (MS) is a chronic disease characterized by multiple demyelinating attacks on the brain or spinal cord. In this chapter, we will discuss the phenotypes of MS, including the most common, relapsing–remitting MS (RRMS), in which there are clinical attacks of distinct neurological symptoms followed by complete or partial improvement. This is the trademark clinical history of MS, and it is accompanied by the characteristic MRI appearance of multiple white matter lesions, with new lesions developing over time. However, another classic feature of the disease is its variability among individuals in the number of relapses and extent of disability progression, which can make it challenging to prognosticate and sometimes to make a firm diagnosis. Fortunately, whereas MS was once solely a clinical diagnosis, MRI has allowed us to identify the disease with more confidence and earlier in the disease’s evolution. We will review the 2010 McDonald criteria, the latest consensus guidelines for diagnosis. We will conclude with a discussion of several conditions that may mimic the symptoms of MS and should be considered early in the diagnostic workup. Ancillary tests, including CSF studies and antiaquaporin-4 neuromyelitis optica (NMO) serum antibody testing, may be helpful where diagnostic dilemmas arise. MS is a primary chronic disease of the central nervous system (CNS) that typically presents in early adulthood. It involves multiple attacks on the brain and/or spinal cord that are separated in space and in time. Sclerosis refers to the plaques or focal areas of demyelination that are the pathological substrate of the disease. The 2010 revised McDonald criteria define an attack as “patient-reported symptoms or objectively observed signs typical of an acute inflammatory demyelinating event in the CNS, current or historical, with duration of at least 24 h, in the absence of fever or infection.” Demyelination may occur in areas that do not give rise to expression of symptoms as well as in neurologically eloquent areas that lead to the attacks or relapses experienced by patients. These silent attacks are most easily appreciated on MRI imaging. Clinical relapses in MS are therefore sometimes referred to as the tip of the iceberg, as there often are considerably more demyelinating lesions that can be visualized on imaging than one would expect from the clinical history. Indeed, one of the classic features of MS is the great variability that exists among affected patients in clinical manifestations, disability progression, and lesion load. The lesions in MS are thought to result from an autoimmune reaction against the CNS. Recent infectious illness and immune activation, such as during the postpartum period, are known to provoke relapses. During an MS relapse, symptoms develop over hours to days then usually persist for weeks before improving. The majority of untreated MS patients experience the occasional clinical relapse every 1–2 years followed by remission with partial or complete recovery (Figure 3.1). This pattern is known as RRMS and affects 85% of those initially diagnosed with MS. Eventually, relapses become less frequent and then cease altogether. Around the same time, disability begins to steadily accumulate. This phase is known as secondary progressive MS (SPMS). There are two other main disease courses in MS: primary progressive MS (PPMS) and progressive relapsing MS (PRMS). Figure 3.1 Disease courses of MS. These drawings illustrate how disability evolves with time in the different courses of the disease. (a) In RRMS, subjects may have complete recovery from intermittent relapses, or they may have partial recovery with some residual disability. (b) Subjects may have progressive disability without relapses in SPMS, or they may have some continuing relapses early in SPMS superimposed on an underlying progressive course. (c) PPMS can progress at a steady rate or may fluctuate in terms of rate of decline, in either event without any clear relapses. (d) In PRMS, there are both relapses and progressive decline from the onset of the disease, and recovery from relapses may be partial or complete. Source: Lublin and Reingold (1996). Reproduced with permission of Lippincott Williams & Wilkins. Declining mobility often helps to mark where RRMS ends and SPMS begins. Over time, patients may progress from ambulating with minor difficulty to needing a cane to walk to using a walker to requiring a wheelchair to being bedbound. Pre-existing deficits, such as impairments in vision, balance, and bladder control, tend to worsen during this stage of the disease. Bulbar dysfunction can occur as an advanced manifestation, and aspiration pneumonia or asphyxia can cause mortality. It can be challenging to define when RRMS transitions into SPMS because relapses may continue even as neurologic disability starts to accumulate. Median time from first symptoms to walking with a cane has been estimated to be between 15 and 30 years with wide variation among studies. More recent studies have shown longer times to walking with a cane, which may reflect benefits from the disease-modifying therapies introduced in the 1990s. By some reports, 15% of people with MS appear to have a more benign or nonprogressive course, even many years after the disease’s onset. About 15% of patients experience an insidious onset of symptoms, with slow but inexorable progression in neurologic impairment. This is the hallmark of PPMS. In the majority of cases, patients present with a spastic paraparesis, often with asymmetric signs. Other common symptoms that may be evident on presentation or develop later include gait difficulties, weakness, ataxia, bladder dysfunction, sexual dysfunction, and sometimes cognitive issues. As in RRMS, it can be helpful to elicit characteristic clinical features like worsening with heat, exercise, and infection. However, history does not reveal episodes of unprovoked acute neurologic worsening. In contrast to RRMS, which predominantly affects women, there is no gender imbalance among those diagnosed with PPMS. Patients are typically about 10 years older at onset compared to RRMS. Disability accumulates more rapidly, but there remains significant variability in the rate of progression as with RRMS. Poor prognostic features include spinal cord lesions, early brainstem or cerebellar lesions, male gender, and younger age at onset. Although MRI findings are similar to RRMS, the cerebral lesion burden and the prevalence of gadolinium-enhancing lesions are typically less than is seen in RRMS and SPMS. MRI is essential in the diagnosis of PPMS as it rules out other important differential diagnoses—structural lesions, cervical spondylopathy, leukodystrophy, and nutritional deficiencies (e.g., vitamin B12 or copper deficiency)—that may be difficult to distinguish on the basis of clinical presentation alone. Approximately 5% of patients present with PRMS. These patients have distinct relapses, in which recovery may be incomplete, after an initial progressive course. Nevertheless, they continue to demonstrate slowly accumulating neurologic disability from disease onset. There is a fifth categorization of MS disease types that is in fact a precursor to MS. The clinically isolated syndrome (CIS) refers to a single demyelinating attack. The majority of patients with CIS will go on to have a second attack, thus meeting criteria for clinically definite MS. A minority will never have another attack. MRI imaging has allowed us to predict the risk of conversion to MS on the basis of multiple white matter lesions in characteristic locations on MRI. In the Optic Neuritis Treatment Trial, there was an overall 50% risk of converting to clinically definite MS 15 years after an isolated optic neuritis. If there were one or more lesions on the MRI, the risk was 72%. The 2010 McDonald diagnostic criteria allow more cases of first demyelinating attacks to be more expeditiously diagnosed as MS. The diagnosis can be made on the basis of a single clinical attack if there is an asymptomatic gadolinium-enhancing lesion and at least one other lesion. Alternatively, a second scan performed at any point following the first that demonstrates a new T2 lesion can be considered as proof of dissemination in time (DIT). Occasionally, a first demyelinating attack can be quite fulminant, and it can be difficult to distinguish between a CIS foreshadowing MS and another pathological demyelinating process such as acute disseminated encephalomyelitis (ADEM) or NMO. The Marburg variant of MS refers to a fulminant form of the disease that is often fatal. Imaging shows large, enhancing lesions with mass effect. Tumefactive MS denotes a large, demyelinating lesion difficult to distinguish from tumor on the basis of imaging alone. It may present in a fulminant manner and be considered as an example of a Marburg variant MS or may present with more focal features. Magnetic resonance spectroscopy can be helpful in making the diagnosis, but sometimes brain biopsy is necessary. A first demyelinating attack can often be quickly identified by its characteristic pattern of onset, the age of the subject, and symptoms involved. MS attacks usually occur in individuals between ages 15 and 50. The onset of symptoms is subacute over several hours to days. Symptoms then typically plateau, although they may persist for weeks to months before improving. Symptoms must persist for at least 24 h to be considered an attack consistent with MS. An attack may consist of one main clinical symptom or multiple symptoms. Occasionally, the development of symptoms will represent a pseudoexacerbation rather than a new lesion. Pseudoexacerbations may be incited by infections, such as a UTI, or by overexertion. A transient worsening or appearance in symptoms with heat is known as Uhthoff’s phenomenon. Patients will often complain of visual blurring, fatigue, or pain during a hot shower or on a very hot day. This finding was first described by Uhthoff in 1890 in the context of worsening of vision in subjects with optic neuritis after exercise. The pathophysiology of Uhthoff’s is likely related to conduction block at elevated temperatures. The threshold for conduction block is lower in demyelinated nerves because of current leakage along the full length of the axon. Common presenting symptoms of MS are listed in Table 3.1. Sensory loss or disturbance is the most common presenting complaint. The most classic description is of numbness, tingling, and pins and needles sensations in the limbs. However, patients may also complain of a limb feeling heavy, cold, swollen, or as if it is vibrating. Some patients describe bilateral limb changes or a band-like sensation around the torso suggesting a spinal cord lesion localization. Spinal cord lesions in MS are often partial and in the posterior cord, with asymmetric examination findings. Table 3.1 Common Presenting Symptoms in MS Visual loss secondary to optic neuritis is the next most common presentation of MS. The affected eye or brow area can be painful, especially with lateral movement. The subject may also complain of photophobia. Color vision is altered, with loss of red–green contrast. Reds are often described as darker or muddier for the impaired eye. There may be a visual field cut, although the field loss is usually central. On examination, the deficit in visual acuity may range from minimal to complete loss of light perception. In general, optic neuritis associated with MS leads to a good recovery in visual acuity. In patients with visual loss that remains fixed, alternative diagnoses should be sought. NMO is another demyelinating disease in which optic neuritis has a much poorer prognosis.
Diagnostic Process
Introduction
Clinical features of MS
Defining MS
MS disease courses
Common symptoms in MS
Most Common Presenting Symptoms
Other Symptoms
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


