Errors in clinical reasoning can lead to a variety of adverse consequences. A common error in diagnostic reasoning is accepting a diagnosis before it is fully verified (premature closure). Kassirer and Kopelman, in a study of diagnostic errors caused by faulty clinical cognition, found that the two most common sources of error lay in faulty triggering (failure to recognize a possible diagnostic hypothesis) and in faulty information gathering and processing. In the latter category, the most common errors related to faulty estimation of disease prevalence (the dictates of Bayes’ theorem) and faulty data interpretation. In a study of common errors in daily practice among neurology residents, the overall rate of diagnostic error and the frequency of diagnostic inaccuracy in various disease entities showed that the initial bedside diagnosis was correct in 67% of patients. The highest rates of inaccuracy were found in the diagnosis of subdural hematoma, myasthenia gravis (MG), subarachnoid hemorrhage (SAH), and Guillain-Barré syndrome (GBS). The common causes of diagnostic inaccuracy were errors of reasoning, an inadequate patient database, and an inadequate fund of knowledge. In a prospective study of the accuracy of bedside diagnoses on a neurology service, patients were evaluated independently by a junior resident, a senior resident, and a staff neurologist. Each individual was required to make an anatomical and etiologic diagnosis based solely on the history and physical examination. In 40 patients with laboratory-confirmed final diagnoses, the clinical diagnoses of the junior residents, senior residents, and staff neurologists were correct in 65%, 75%, and 77%, respectively. The errors by the junior residents, <senior residents>, and (staff) were attributed to incomplete history and examination in 4 <1> (0), inadequate fund of knowledge in 4 <3> (3), and poor diagnostic reasoning in 6 <6> (6). Thus, experience beyond a certain level is not necessarily a cure for faulty reasoning. Vickery et al. presented five common sources of cognitive diagnostic error: framing effects, anchoring, the availability heuristic, pattern misrecognition, and blind obedience and overreliance on test results without considering sensitivity and specificity.
NEUROLOGIC DIFFERENTIAL DIAGNOSIS
Pathologic processes behave in certain ways depending on their location in the nervous system, and in certain other ways related to their inherent natures. Neurologists deal in two basic clinical exercises: Where is the lesion in the nervous system and what is the lesion in the nervous system or differential diagnosis by location and differential diagnosis by pathophysiology or etiology. The anatomic diagnosis and the etiologic diagnosis aid and support each other. In general, the neurologic examination aids primarily in establishing the anatomic or localization diagnosis and the history aids in the etiologic diagnosis, but there is overlap. The examination also serves to indicate the severity of the abnormality. A dependence on neuroimaging and other tests as the primary approach to diagnosis causes many errors. Defining the patient’s illness first in terms of anatomy and likely etiology helps insure the appropriate use of neurodiagnostic studies.
Pathophysiologically, disease of the nervous system may cause manifestations due to destruction, release, “irritation,” and partial assumption of function by healthy tissues. Symptoms and signs of destruction result from a transient or permanent loss of function similar to the manifestations of disease commonly found in other body systems. Peripheral nerve injury causes weakness, sensory loss, and areflexia in the distribution of the nerve. Destruction of the cerebral cortex may cause paresis, hypesthesia, blindness, or intellectual loss. Symptoms related to release of function occur when there is disease of a portion of the nervous system that has an inhibitory function. Some responses may be exaggerated because of disinhibition and the release of intact centers from higher control. A lesion of the corticospinal system is often followed by increased muscle tone, increased reflexes, and the presence of certain pathologic reflexes. These are positive rather than negative signs of disturbed function. Positive signs are apparent in a different manner in the presence of overactivity, excitation, or “irritation” of a part of the nervous system. Characteristic examples are the pain and muscle spasm that follow disease of a peripheral nerve, and convulsions causing increased motor activity. Partial assumption of function by healthy tissues may compensate for loss of function due to disease in another part. There is a certain amount of overlapping and duplication of function in the nervous system; an intact center, nerve, or muscle may assume some of the physiologic activity of a diseased part. In some parts of the nervous system, large lesions may cause a paucity of signs and symptoms because of a minimum of physiologic activity of the part, duplication of function, or compensation elsewhere.
The first diagnostic consideration is whether the patient has an organic disease or whether the symptoms are likely psychogenic. If the disorder is organic, consider whether the condition is a primary neurologic disease, a neurologic complication of a systemic disorder, a neurologic complication of drug or medication use, or the effects of a toxin.
ANATOMICAL DIAGNOSIS
The patterns of abnormality help to localize a disease process to a particular part of the nervous system. Clinical features that are particularly helpful in neurologic differential diagnosis include the distribution of any weakness; the presence or absence of sensory symptoms; the presence or absence of pain; the presence or absence of cranial nerve abnormalities and whether they are ipsilateral or contralateral to the other abnormalities on examination; the status of the reflexes; the presence of pathologic reflexes; involvement of bowel and bladder function; and the presence or absence of symptoms that clearly indicate cortical involvement, such as seizures, aphasia, or altered mental status (AMS). Weakness may be unilateral or bilateral, symmetric or asymmetric, primarily proximal or primarily distal; each of these patterns has differential diagnostic significance. The pattern of sensory abnormalities also provides significant information.
It may be helpful to organize the nervous system anatomically by considering sequentially more peripheral or central structures, beginning either at the cerebral cortex or the muscle. Consider each level where disease tends to have a characteristic and reproducible clinical profile. At each major level, disease processes tend to have characteristic clinical features, although with some degree of overlap. For example, disease involving the muscle, neuromuscular junction (NMJ), peripheral nervous system (PNS), nerve roots, spinal cord, brainstem, and hemispheres each tend to produce a characteristic clinical picture. These are discussed in the following chapters. Most of these levels can be further subdivided to narrow the differential diagnosis. Some diseases cause multifocal or diffuse abnormalities, and these are often particularly challenging. By trying to localize the disease process to one or two likely levels, such as muscle or NMJ, one can think more systematically about the etiologic possibilities. Manifestations of disease in different locations can sometimes be similar, causing confusion. Extraocular muscle dysfunction due to MG can simulate an internuclear ophthalmoplegia. Deafferentation due to peripheral nerve or posterior column disease can cause sensory ataxia difficult to distinguish from cerebellar disease. Distinguishing early GBS from early transverse myelopathy can be difficult.
Assuming the process under consideration is indeed neurologic, the first major attempt at localization should be to decide if the pathologic process likely lies in the PNS or the central nervous system (CNS). For purposes of this discussion, the PNS includes the NMJ and the muscle. Clinical features helpful in distinguishing PNS disease from CNS disease are summarized in Table 53.2. Some neurologic signs and symptoms occur very commonly in both CNS and PNS diseases, and arriving at the correct diagnosis sometimes depends more on analyzing the associated findings than the presenting manifestation. Some of the abnormalities that occur commonly in both peripheral and central disease include abnormal pupils, ptosis, diplopia, dysphagia, dysarthria, weakness, sensory loss, and difficulty walking.
TABLE 53.2 Clinical Features That Suggest Central (Brain and Spinal Cord) versus Peripheral (Roots, Plexi, Peripheral Nerves, Neuromuscular Junctions, and Muscle) Nervous System Disease

CK, creatine kinase; EEG, electroencephalogram; EMG, electromyogram; MRI, magnetic resonance imaging.
CLINICAL MANIFESTATIONS OF DISEASE
The following paragraphs provide a brief summary of the clinical manifestations of disease at different levels of the nervous system, from muscle to cerebral cortex.
Myopathy
Myopathies are those conditions in which there is a primary dysfunction of skeletal muscle. Patients with muscle disease usually have symmetric, proximal weakness. They have trouble getting up from a chair, difficulty getting out of a car, and difficulty raising their arms overhead. Patients often have difficulty with everyday grooming activities, such as shaving or handling hair and makeup. Early on, patients with acquired muscle disease switch from tub baths to showers because they cannot get out of the tub. The examination typically reveals proximal weakness reflecting the clinical complaint. Reflexes are usually preserved unless the muscle weakness is very severe. There are no pathologic reflexes. There is no sensory loss; bowel and bladder dysfunction generally do not occur; and there are no defects in coordination, mentation, or higher cortical function. The gait in myopathy is often abnormal, characteristically with a waddling character. Atrophy is not prominent unless the process is severe, as in some dystrophies. Pseudohypertrophy may occur in dystrophies, particularly dystrophinopathies. Patients may or may not have muscle pain, tenderness, or soreness, but usually they do not. Some muscle disorders are accompanied by myotonia. Many muscle diseases can cause rhabdomyolysis and myoglobinuria.
Although most myopathies cause symmetric proximal weakness, some conditions cause weakness with atypical patterns. The weakness in myotonic dystrophy is predominantly distal. The distal myopathies are a group of conditions, mostly hereditary, that produces primarily distal weakness. A pattern of weakness involving the proximal upper extremities, particularly the periscapular region, and distal lower extremities occurs in the scapuloperoneal syndromes, which may be either myopathic or neuropathic. Weakness of the proximal lower extremities, particularly the quadriceps, and the distal upper extremities, particularly the wrist and finger flexors, occurs in inclusion body myositis.
The causes of myopathy are legion (Table 53.3). Myopathies can be divided into those that are inherited and those that occur sporadically, as this is often the initial step in clinical differential diagnostic thinking. A partial list of etiologies includes inflammatory myopathies, muscular dystrophies, congenital myopathies, metabolic and mitochondrial myopathies, toxic myopathies, and myopathies arising as a complication of many different systemic disorders.
TABLE 53.3 Common Primary Muscle Diseases

HTLV-1, human T-cell lymphotrophic virus type 1; MELAS, mitochondrial encephalomyopathy, lactate acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fiber disease.
Neuromuscular Junction Disorders
The cardinal manifestation of diseases involving the NMJ is weakness due to impaired neuromuscular transmission (NMT). The character and distribution of the weakness and associated manifestations vary among the different conditions. The most common conditions encountered clinically are MG and the Lambert-Eaton myasthenic syndrome (LEMS). However, the most common condition by far is MG. Table 53.4 summarizes the clinical characteristics of these two conditions. Other rare disorders that can cause clinically significant NMT disorders include botulism, hypermagnesemia, and exposure to some toxins.
TABLE 53.4 Comparison between Myasthenia Gravis (MG) and Lambert-Eaton Syndrome
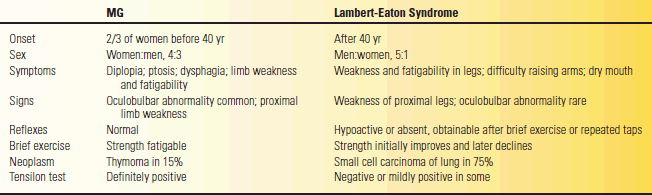
Modified from Oh SJ. Electromyography: Neuromuscular Transmission Studies. Baltimore: Williams & Wilkins, 1988.
Patients with NMJ disorders usually have symmetric, proximal muscle weakness, which can simulate a myopathy, but in addition often have bulbar involvement. The weakness is typically fatigable; it varies and fluctuates with the level of activity and with the time of day. Most commonly, patients have weakness of eye movement causing double vision or ptosis of one or both eyelids. They may have trouble talking and swallowing, with a tendency to nasal regurgitation of fluids. Such symptoms and signs of bulbar weakness are one of the main differences between an NMJ disorder and a myopathy. There is no pain or sensory loss. Deep tendon reflexes (DTRs) are normal in MG but may be depressed in LEMS and other presynaptic disorders. There are no pathologic reflexes.
In the typical case of MG, weakness prominently involves the eyelids and extraocular muscles, resulting in fluctuating ptosis and diplopia, which varies with the time of day and with activity of the muscles. The ptosis and diplopia are frequently less severe in the morning and grow worse as the day wears on. Ptosis not present at rest can often be elicited by sustained upgaze, which fatigues the eyelid levators. When involvement is limited to the extraocular muscles, eyelids, and orbicularis oculi, the condition is termed ocular or purely ocular myasthenia. With some patients, the disease may never progress beyond this point. In most, generalized myasthenia eventually develops, with eye symptoms remaining prominent. The differential diagnosis of MG includes many neuromuscular and nonneuromuscular conditions. A minimal list includes thyroid eye disease, brainstem disease, mitochondrial myopathy, inflammatory myopathy, oculopharyngeal dystrophy, motor neuron disease, and cranial nerve compressive lesions.
Peripheral Neuropathy
Peripheral neuropathies are conditions that affect peripheral nerve axons, their myelin sheaths, or both. The cardinal manifestations of peripheral neuropathy are weakness, alterations in sensation, and reflex changes. Common causes of peripheral neuropathy include diabetes mellitus, alcoholism, and GBS. Patients with generalized polyneuropathy have symmetric, predominantly distal weakness, sensory loss, depressed or absent DTRs, no pathologic reflexes, and no bowel or bladder dysfunction. Pain is a common accompaniment and often a major clinical feature. While it is a good general rule that muscle disease causes proximal weakness and generalized peripheral nerve disease causes distal weakness, there are exceptions. For instance, myotonic dystrophy is a common muscular dystrophy that produces distal weakness, and GBS is a common peripheral nerve disease that frequently produces proximal weakness.
Peripheral nerve diseases are divided into polyneuropathies (all the nerves are affected) and mononeuropathies. In multiple mononeuropathy (mononeuritis multiplex), more than one nerve is affected but not all. With a mononeuropathy, symptoms and signs are specifically related to the affected nerve (see Chapter 46). Table 53.5 lists some of the etiologies of peripheral nerve disease but is far from exhaustive. Nerve fibers react to injury through two primary mechanisms: axonal degeneration and demyelination. In axonal neuropathies the primary pathology is degeneration of the axonal cytoplasm. Wallerian degeneration specifically refers to the axonal degeneration distal to a traumatic nerve injury. In demyelinating neuropathies, the primary insult is to the myelin sheath or the Schwann cell. The ultimate goal in neuropathy evaluations is to establish a precise etiologic diagnosis in order to guide treatment, if any is available. The most important exercise is to distinguish demyelinating neuropathy from axonopathy (Table 53.6). In compression neuropathies, the demyelination is focal and involves only a discrete segment of nerve (segmental demyelination, e.g., carpal tunnel syndrome).
TABLE 53.5 Some Causes of Peripheral Neuropathy

Modified from Campbell WW. Essentials of Electrodiagnostic Medicine. Philadelphia: Lippincott Williams & Wilkins, 1999.
TABLE 53.6 Clinical Features That Help Distinguish Axonopathy from Myelinopathy
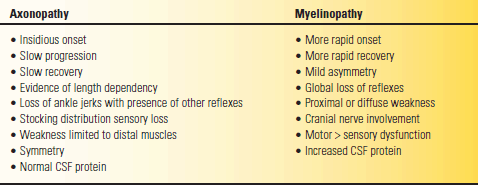
CSF, cerebrospinal fluid.
Modified from Campbell WW. Essentials of Electrodiagnostic Medicine. Philadelphia: Lippincott Williams & Wilkins, 1999.
Plexopathy
Diseases involving the brachial plexus are much more common than those involving the lumbosacral plexus. Patients with plexus disorders have clinical deficits that mirror the involved structures, so a knowledge of plexus anatomy is vital to deciphering the deficit. There is typically both weakness and sensory loss involving all or part of an extremity, accompanied by depressed or absent DTRs in the involved area, no pathologic reflexes, and no bowel or bladder dysfunction. Other neurologic functions are intact. Plexopathy is discussed in more detail in Chapter 46.
Radiculopathy
Most radiculopathies are due to disc herniations or spondylosis. When severe, there are both motor and sensory deficits and a depressed DTR in the distribution of the involved root(s). Pain is common and often severe, usually accompanied by pain and limitation of motion of either the neck or lower back, along with signs of root irritability, such as a positive straight leg raising test. There are no pathologic reflexes, and no bowel or bladder dysfunction. The presence of these findings suggests there is concomitant spinal cord compression. Radiculopathy is discussed in more detail in Chapter 47.
Myelopathy
Spinal cord disorders often produce characteristic patterns of clinical abnormalities, with motor and sensory deficits in a certain distribution. Commonly seen patterns include transverse myelopathy, the Brown-Séquard (hemicord) syndrome, anterior cord syndrome, central cord syndrome, a syringomyelic pattern, posterolateral sclerosis or combined system disease, and an anterior horn syndrome (see Chapter 24). With transverse myelopathy, there is symmetric involvement causing bilateral weakness below a particular level, producing either paraparesis or quadri-paresis. In addition to weakness below the level of the lesion, patients with spinal cord lesions may also have paresthesias, numbness, tingling, and sensory loss with a discrete sensory level, usually on the trunk. Common patterns of sensory loss are summarized in Table 53.7. The pattern of weakness is typically more localizing than sensory abnormalities in lesions of the cervical spinal cord, while the demonstration of a sensory level on the trunk is more helpful in localizing lesions of the thoracic cord.
TABLE 53.7 Findings on Sensory Examination That Are Strongly Suggestive of a Lesion of the Spinal Cord or Cauda Equina

Modified from Woolsey RM, Young RR. The clinical diagnosis of disorders of the spinal cord. Neurol Clin 1991;9:573–583.
Common causes of myelopathy include compression, trauma, and acute transverse myelitis. Spinal cord mass lesions are most commonly due to tumor, most often metastatic, abscess, or disc herniation. The lesion may be intramedullary, within the cord substance, or extramedullary, compressing the spinal cord or its blood supply. Table 53.8 summarizes some of the clinical features that help distinguish between extramedullary and intramedullary lesions. An extramedullary lesion may be intradural or extradural. Extradural tumors are generally malignant and the intradural tumors benign. A long duration of symptoms is more consistent with an intradural lesion. Other important causes of myelopathy include retroviral infection—HIV and human T-cell lymphotrophic virus type 1—as well as connective tissue disorders, mucopolysaccharidosis, neurosarcoidosis, and radiation therapy.
TABLE 53.8 Signs and Symptoms Differentiating between Extramedullary and Intramedullary Tumors of the Spinal Cord

Brainstem Disease
The classic distinguishing feature of brainstem pathology is that deficits are “crossed,” with cranial nerve dysfunction on one side and a motor or sensory deficit on the opposite side. Common causes of brainstem disease include stroke, MS, and neoplasm. There are often symptoms reflecting dysfunction of other posterior fossa structures, such as vertigo, ataxia, dysphagia, nausea and vomiting, and abnormal eye movements. Unless the process has impaired the reticular activating system, patients are normal mentally, awake, alert, able to converse (though perhaps dysarthric), not confused, and not aphasic. The DTRs are usually hyperactive with accompanying pathologic reflexes in the involved extremities; pain is rare and sphincter dysfunction occurs only if there is bilateral involvement. Brainstem disorders are discussed further in Chapter 21.
Cranial Neuropathy
Disease may selectively involve one, or occasionally more than one, cranial nerve. The long tract abnormalities, vertigo, ataxia, and similar symptoms and findings that are otherwise characteristic of intrinsic brainstem disease are lacking. Common cranial neuropathies include optic neuropathy due to MS, third nerve palsy due to aneurysm, and Bell’s palsy. Involvement of more than one nerve occurs in conditions such as Lyme disease, sarcoidosis, and lesions involving the cavernous sinus. Cranial neuropathies are discussed further in Chapters 12 to 21.
Cerebellar Disease
Patients with cerebellar dysfunction suffer from various combinations of tremor, incoordination, difficulty walking, dysarthria, and nystagmus, depending on the parts of the cerebellum involved. There is no weakness, sensory loss, pain, hyperreflexia, pathologic reflexes, sphincter dyscontrol, or abnormalities of higher cortical function. When cerebellar abnormalities result from dysfunction of the cerebellar connections in the brainstem, there are usually other brainstem signs. Cerebellar disorders are discussed further in Chapter 43.
Basal Ganglia Disorders
Diseases of the basal ganglia cause movement disorders such as Parkinson’s disease (PD) or HD. Movement disorders may be hypokinetic or hyperkinetic, referring to whether movement is in general decreased or increased. PD causes bradykinesia and rigidity. Huntington’s disease, in contrast, causes increased movements, which are involuntary and beyond the patient’s control (chorea). Tremor is a frequent accompaniment of basal ganglia disease. Basal ganglia disorders are discussed further in Chapters 26 and 30.
Cerebral Hemisphere Disorders
Characteristic of unilateral hemispheric pathology is a “hemi” deficit: hemisensory loss, hemiparesis, hemianopsia, or perhaps hemiseizures. Other common manifestations include hyperreflexia and pathologic reflexes. Pain is not a feature unless the thalamus is involved, and there is no difficulty with sphincter control unless both hemispheres are involved. Within this framework, disease affecting the cerebral cortex behaves differently from disease of subcortical structures. Patients with cortical involvement may have aphasia, apraxia, astereognosis, impaired two-point discrimination, memory loss, cognitive defects, focal seizures, or other abnormalities that reflect the essential integrative role of the cortex. Processes affecting the dominant hemisphere often cause language dysfunction in the form of aphasia, alexia, or agraphia. With disease of the nondominant hemisphere, the patient may have higher cortical function disturbances involving functions other than language, such as apraxia. If the disease affects subcortical structures, the clinical picture includes the hemidistribution of dysfunction but lacks those elements that are typically cortical (e.g., language disturbance, apraxia, seizures, dementia). Certain processes involve wide areas of the cerebrum, causing diffuse dysfunction. In addition, some disorders may produce increased intracranial pressure, which creates additional abnormalities due to edema and pressure superimposed on the clinical manifestations related to the underlying pathology.
Multifocal/Diffuse Disorders
Some disease processes are diffuse or multifocal, producing dysfunction at more than one location, or involve a “system.” For example, neuromyelitis optica characteristically affects both the spinal cord and the optic nerves (i.e., it is multifocal). Amyotrophic lateral sclerosis is a system disorder causing diffuse dysfunction of the entire motor system from the spinal cord to the cerebral cortex, sparing sensation and higher cortical function.
Disorders of the Meninges, Ventricular System, and Intracranial Pressure
Many conditions can affect the meninges, including infections, neoplasia, sarcoidosis, and others. The most common disorders are infectious and present with evidence of infection and increased intracranial pressure. Some meningeal infections may be extremely indolent and lack the classical signs associated with infection. Chronic meningitis can also present as dementia or AMS. Abnormalities of the ventricular system can occur due to congenital anomalies, such as aqueductal stenosis, or acquired conditions, such as normal pressure hydrocephalus. Dilatation of the ventricular system may cause head enlargement in children. In adults, such conditions usually present with evidence of increased intracranial pressure or with dementia, AMS, gait problems, or difficulty with bladder control.
Disorders of the Skull and Vertebral Column
Disorders of the skull and vertebral column range from the mundane and minimally significant, such as spina bifida occulta, to the horrific, such as disfiguring craniosynostosis syndromes. The most common conditions are due to trauma, such as skull or spinal fractures. Occasional patients with bony tumors may present with localized pain. Sometimes bony lesions are picked up as incidental findings on radiographic studies done for other reasons. Congenital and developmental skeletal disorders may be immediately obvious at birth, such as myelomeningocele, or present well into adulthood, such as spinal dysraphism. Conditions may be limited to a bony abnormality, such as a linear skull fracture or spondylolysis, or involve neural structures as well, such as a depressed skull fracture or diastematomyelia. In the absence of trauma, the challenge is to remember to consider the possibility of a congenital or developmental skull or spinal disorder, even in the adult patient.
Disorders of the Vascular System
Diseases affecting the vascular system typically present as an ischemic or hemorrhagic event, single or multiple. The usual clue to a vasculopathy is multiple events involving different parts of the nervous system. Other presentations, such as dementia, can occur as well. Rare but important nonatherosclerotic conditions include vasculitis, Moya-moya angiopathy, arterial trauma and dissections, fibromuscular dysplasia, migraine, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, amyloid angiopathy, and complications of radiation therapy. Cerebrovascular disease may complicate systemic conditions such as collagen vascular disease and hypertensive encephalopathy. Hematologic disorders such as sickle cell disease, thrombotic thrombocytopenic purpura (TTP), polycythemia, intravascular lymphoma, and antiphospholipid syndrome may cause stroke. Coagulopathies may cause bleeding, as from anticoagulant effects or disseminated intravascular coagulation. The most common condition causing hypercoagulability-related stroke is activated protein C resistance. Vasculitis may complicate infections such as meningitis, meningovascular syphilis or herpes zoster. Cerebral vasculitis may complicate systemic vasculitis, such as in systemic lupus, polyarteritis, Wegener’s granulomatosis, and Churg-Strauss syndrome. Isolated angiitis of the brain may cause both infarct and hemorrhage. Other vasculitic disorders include Behcet’s disease, Susac’s syndrome, and Sneddon’s syndrome.
DIFFERENTIAL DIAGNOSIS BY ETIOLOGY
From a differential diagnostic standpoint, it is usually most helpful to think first about the localization of the disease process in the nervous system, and secondarily about the etiology. Localization limits the etiologic differential diagnosis since certain disease processes typically involve or spare particular structures. If the clinical manifestations suggest cerebellar disease, then muscular dystrophy, MG, and GBS are not diagnostic considerations. Knowing the likely location of the pathology generally places the condition into a broad etiologic differential diagnostic category. Occasionally, the etiology is very obvious, such as stroke or CNS trauma, and the diagnostic exercise focuses mostly on the localization.
Categories and etiologic classifications of neurologic disease are necessarily somewhat arbitrary, as is the category in which to place a given entity. For instance, subacute combined degeneration of the spinal cord could be seen as a metabolic disorder, a nutritional deficiency, or as a complication of a systemic illness, pernicious anemia. Any classification scheme is stressed in an age in which we recognize entities as diverse as high-altitude cerebral edema (HACE), ciguatera intoxication, critical illness myopathy, sleep apnea, mitochondropathies, Whipple’s disease, intracranial hypotension, Susac’s syndrome, NMDA encephalitis, Hashimoto’s encephalopathy, and the jumping Frenchmen of Maine as conditions neurologists should be familiar with. Porphyria is one example of many conditions that are both metabolic and genetic disorders. The etiologic classifications used here are neoplasm, vascular disease, infection, inflammatory and autoimmune disorders, trauma, pharmaceuticals and other chemical agents, substance-related disorders, toxins, metabolic disorders, demyelinating disease, congenital and developmental abnormalities, genetic disorders, degenerative conditions, disorders due to physical agents, environmental related conditions, mitochondropathies, channelopathies, paroxysmal disorders (seizures, headache and sleep), complications of systemic conditions, and nonorganic and psychiatric disease. Conditions such as epilepsy and migraine have important clinical manifestations far beyond the individual seizure or headache. The following paragraphs briefly summarize the features of some of these etiologic categories.
Neoplasms
Neoplasms may be divided into those that are intra-axial, arising within the brain or spinal cord substance, and those that are extra-axial, involving the meninges, cranial nerves, and other surrounding structures. Tumors are named according to their resemblance to cells that are found in the normal mature and developing nervous system. Intra-axial tumors are composed of cells of neuroectodermal origin. Examples of common intra-axial tumors include astrocytomas, oligodendrogliomas, ependymomas, medulloblastomas, and primary brain lymphoma. All are malignant because they invade the substance of the brain, but some more so than others. Common extra-axial tumors include pituitary adenoma, acoustic neuroma, and meningioma. Extra-axial tumors are more likely to be histologically benign and amenable to excision. They produce neurologic dysfunction primarily by exerting pressure rather than by invading. The symptoms and signs of intracranial tumors depend on their location, mass effect, pathologic characteristics, as well as their tendency to cause an increase in intracranial pressure. Focal manifestations of intracranial tumors include irritative phenomena such as seizures and symptoms of destruction such as progressively severe dysfunction of the structures involved.
Metastatic tumors are the most common type of intracranial neoplasm. Of the primary intracranial neoplasms, approximately 50% belong to the glioma group. Astrocytoma is the most common primary intracerebral neoplasm. Meningiomas make up about 15% of all intracranial neoplasms. Most are supratentorial and produce symptoms and signs by pressure on the brain. An acoustic neuroma (schwannoma, neurinoma, neurilemmoma) is a tumor that usually arises from the vestibular portion of the eighth cranial nerve; it is by far the most common tumor to involve cranial nerves. Pituitary adenomas are relatively common and may cause both endocrine disturbances as well as neurologic dysfunction because of mass effect. Craniopharyngiomas are tumors of congenital origin that arise from cell rests in the region of the pituitary. They usually become manifest in childhood or young adult life.
Malignancies can produce a host of nonmetastatic, paraneoplastic neurologic syndromes. The neurologic syndrome may precede the malignancy by months or years, occur simultaneously with presentation of the tumor, or develop in patients with known cancer. The paraneoplastic syndromes include but are not limited to progressive cerebellar ataxia, peripheral neuropathy, Lambert-Eaton syndrome, opsoclonus-myoclonus syndrome, “limbic encephalitis” (memory and emotional disturbances), and sensory ataxia due to dorsal root ganglion cell degeneration.
Vascular Disease
Stroke remains the third leading cause of death in the United States. Recognized risk factors are increasing age, hypertension, diabetes mellitus, dyslipidemia, history of transient ischemic attack (TIA), cigarette smoking, carotid artery stenosis, and heart disease. Generally, cerebrovascular disease can be broadly divided into ischemic and hemorrhagic types (Table 53.9). It can be classified in a number of other clinically relevant ways: anterior circulation (carotid) versus posterior circulation (vertebrobasilar), large vessel (atherosclerotic) versus small vessel (hypertensive, diabetic, lacunar), and thrombotic versus embolic. Patients may present with many varieties of stroke related to these variables, as well as to the particular location of the event in the brain. About 70% to 80% of intracranial vascular events are ischemic, 15% are primary intracerebral hemorrhage (ICH), and about 5% are SAH. The majority of initial ischemic strokes are atherothrombotic infarctions; cardiac embolism produces about 15% to 30% of cases, small vessel lacunar disease about 15% to 30%, and other types, such as vasculitis or arterial dissection, 3%.
TABLE 53.9 A Classification Scheme for Stroke

From Campbell WW, Pridgeon RP. Practical Primer of Clinical Neurology.
Philadelphia: Lippincott Williams & Wilkins, 2002.
Ischemic disease tends to present with an acute focal deficit in an alert patient. Intracranial hemorrhage is more likely to have an apocalyptic onset with coma and a poor outcome. Ischemic cerebrovascular disorders can be further subdivided into those due to thrombotic occlusion of a vessel, those due to embolic occlusion from distant sources such as the heart or the great vessels in the neck and chest, watershed infarcts occurring at border zones of perfusion when there is a general decrease in cerebral perfusion, lacunar infarcts (less than 2 cm in diameter and usually deep, the result of occlusion of perforating “end” arteries), and TIA. The onset of symptoms with all of these is usually abrupt, although the symptoms of thrombosis occasionally appear more gradually than those of either hemorrhage or embolism. It is important to identify precise time of onset, since this determines eligibility for treatment with tissue plasminogen activator, which must be given within 3 hours of symptom onset.
Stenosis or occlusion of extracerebral and even extracranial arteries is responsible for a large proportion of cerebrovascular disease. Affected vessels may be in the neck or chest and include the common and internal carotids, the vertebrals, and the arch of the aorta. Small vessel disease involves deep, penetrating small arteries and arterioles and is frequently related to hypertension. Large vessel disease tends to present as TIA or cortical stroke, small vessel disease as a sub-cortical lacunar syndrome. Anterior circulation events typically produce hemispheric infarction causing hemiparesis and higher cortical function defects such as aphasia, whereas posterior circulation events cause brainstem or occipital lobe ischemia. Thrombotic events tend to have onset during sleep and cause less severe, more restricted deficits. Embolic events classically occur during activity, are more devastating, and are more likely to have associated cardiac disease.
Patients with TIAs experience brief episodes of neurologic dysfunction, by traditional definition lasting less than 24 hours but in fact usually lasting only 10 to 30 minutes and rarely more than 1 hour. The attacks resolve to leave no detectable clinical deficit. Three basic forms are recognized: carotid distribution TIA (brief hemispheric spells), vertebrobasilar TIA (brainstem ischemia or visual field deficits), and amaurosis fugax (transient monocular visual disturbances due to ischemia in the ophthalmic artery distribution). TIAs presage major stroke in about 25% to 30% of patients. In a patient with TIA, the stroke risk is about 5% to 6% per year for the first 5 years and is greatest in the first year.
In ischemic stroke, the deficit depends on the arterial territory involved. Anterior cerebral artery ischemia is characterized by disproportionate weakness and numbness of the contralateral leg. Areas of particular clinical importance perfused by the middle cerebral artery (MCA) include the frontal eye fields, Broca’s area, Wernicke’s area, and the cortical areas subserving motor and sensory function for the arm and face. Large infarctions involving the entire MCA territory of the dominant hemisphere typically cause contralateral hemiplegia, hemianesthesia, dense homonymous hemianopsia, and global aphasia. Complete MCA distribution lesions in the nondominant hemisphere produce hemiplegia, various forms of apraxia, a visual field deficit, and differing combinations of the peculiar syndrome of neglect of the left side of space, denial of disability, and sometimes total failure to recognize the paralyzed extremities as part of the body (anosognosia). Patients with posterior cerebral artery strokes typically have homonymous hemianopia as the predominant clinical manifestation and may have no significant weakness or sensory loss. Internal carotid artery (ICA) occlusion may cause infarction of the entire hemisphere with the exception of the thalamus, inferior portion of the temporal lobe, and medial portion of the occipital lobe. Large infarctions, such as those that occur with ICA or MCA occlusions, often cause significant cerebral edema. Of all fatal ischemic strokes, cerebral edema and increased intracranial pressure are the cause of death in about one-third. Brainstem strokes are characterized by “crossed” syndromes of cranial nerve dysfunction ipsilateral to the lesion and long motor or sensory tract dysfunction contralaterally.
Many strokes are due to lacunar infarction related to fibrinoid necrosis, or lipohyalinosis, of small arterioles throughout the body. Hypertension is responsible for about 80% to 90% of lacunar infarctions. Diabetes mellitus is another important predisposing condition. Lacunar infarcts primarily affect subcortical structures such as the basal ganglia, thalamus, internal capsule, subcortical white matter, cerebellum, and brainstem. They do not involve the cerebral cortex. Occlusion of these small endarteries produces infarction, and the small infarctions result in little cavities filled with fluid (Fr. lacune “lake”), from 2 to 15 mm in size. The symptoms that occur depend on the location, but diffusion-weighted magnetic resonance imaging (MRI) studies have shown that the same lacunar syndrome can result from a lesion in a variety of locations and that lesions in the same location can cause different lacunar syndromes. There are four classical lacunar strokes: pure motor stroke (PMS), pure sensory stroke, dysarthria-clumsy hand syndrome, and ataxic hemiparesis. Many other lacunar syndromes have been recognized; the present count is over 20. PMS is the most common and clinically best characterized of the lacunar syndromes. It accounts for about 10% of patients with acute stroke, and 50% of patients with lacunar stroke. The lesion usually involves the posterior limb of the internal capsule, damaging the corticospinal tract fibers in isolation and causing a dense hemiparesis but no sensory loss, visual field deficit, speech disturbance, eye movement disorder, or other evidence of dysfunction of the cerebral cortex—a pure motor deficit. The other lacunar syndromes are much less common than PMS.
In contrast to ischemic cerebrovascular disease, intracranial hemorrhage characteristically produces either severe headache or early impairment of consciousness, or both. Intracranial hemorrhage may occur into the parenchyma or into one of the spaces that surround the brain. Intraparenchymal bleeding may occur into the supratentorial compartment (intracerebral), the cerebellum, or the brainstem. Supratentorial hemorrhage is often further divided into basal ganglia (usually putaminal) hemorrhage, thalamic hemorrhage, and so-called lobar or subcortical hemorrhage, which involves the deep white matter in the corona radiata. Extraparenchymal hemorrhage may involve the subarachnoid, subdural, or epidural spaces. Most extraparenchymal hemorrhage is due to head trauma. Spontaneous intracranial, extraparenchymal hemorrhage is usually into the subarachnoid space.
Hypertensive ICH usually involves the basal ganglia, subcortical white matter, thalamus, pons or cerebellum, with basal ganglia and thalamic bleeds accounting for the vast majority. Patients typically have apocalyptic events with dense deficits and rapid impairment of consciousness. Trauma is the most frequent cause of SAH. Nontraumatic SAH is most often due to ruptured saccular aneurysm, occasionally to arteriovenous malformation, and in 10% to 15% of the cases to no identifiable etiology. The major etiologies of aneurysms are congenital, atherosclerotic, mycotic, and dissecting. Most saccular (berry) aneurysms occur at branching sites of the major arteries of the circle of Willis. The etiology is a combination of congenital and acquired factors. About 80% of berry aneurysms involve the anterior circulation and about 20% are located in the vertebrobasilar system. The most common sites of intracranial berry aneurysms are the distal ICA, posterior communicating artery, anterior communicating artery, tip of the basilar artery, middle cerebral bifurcation, and the posterior inferior cerebellar artery. Posterior communicating artery aneurysms frequently compress the oculomotor nerve. With aneurysmal SAH, the onset is usually precipitous. The patient develops a sudden, severe headache, often occipital or nuchal (thunderclap headache), often accompanied by convulsions, obtundation, or coma. Other causes of intracranial hemorrhage include amyloid angiopathy, vasculitis, and mycotic aneurysm.
Most cerebrovascular disease is related to atherosclerosis and hypertension, but there are other important etiologies (see above). Occlusive disease of the cerebral veins and the venous sinuses may also occur.
Intracranial Infections
Numerous pathogenic microorganisms can infect the CNS. Clinical manifestations depend on the nature of the infecting organism, adequacy of host defenses, and the CNS area predominately involved. The clinical course may range from hyperacute (meningococcal meningitis), to chronic (tuberculous meningitis), to extremely chronic (prion infection). Many infections once rare have become commonplace since the advent of AIDS.
In acute bacterial meningitis, the patient typically appears acutely ill and toxic with fever, headache, altered sensorium, and stiff neck. Low sugar and polymorphonuclear leukocytosis characterize the cerebrospinal fluid (CSF). The differential diagnosis depends greatly on age and circumstances. Between 19 and 59 years, most cases are due to Streptococcus pneumoniae, and the next most common etiology is Neisseria meningitidis; over the age of 60, the most common organisms are S. pneumoniae and Listeria monocytogenes. The possibility of bacterial meningitis should be considered in any patient with headache or AMS accompanied by fever. Patients with viral aseptic meningitis present in much the same way as patients with bacterial meningitis, except they generally appear less sick. The CSF in viral meningitis contains a predominance of mononuclear cells, a normal sugar level, and variable protein elevation. Many different viruses can produce aseptic meningitis. Other causes of an aseptic meningeal syndrome include neoplastic invasion of the meninges, reaction to certain medications, chemically induced meningeal inflammation, and infection by organisms difficult to culture. The term aseptic also applies to these forms of meningitis since routine bacteriologic cultures prove sterile, but aseptic is often used synonymously with viral meningitis. Some organisms can produce an indolent form of meningitis. Major considerations include tuberculosis, cryptococcosis and other fungi, Lyme disease, and sarcoidosis. The CSF sugar is frequently low but rarely as low as in bacterial meningitis. Protein elevations, sometimes striking, are the rule. Brain abscesses can arise either because of direct spread from a contiguous infected source, such as a mastoid, or because of hematogenous spread. Patients typically present with varying combinations of headache, progressive neurologic deficits, seizures, and evidence of infection. However, fever and leukocytosis are absent in about half of the patients harboring a brain abscess.
Viral encephalitis differs from viral meningitis by virtue of the involvement of the brain parenchyma, which may produce altered consciousness progressing to coma, seizures, or focal signs such as hemiparesis, visual field deficits, and aphasia. Meningoencephalitis refers to involvement of both the meninges and the parenchyma. The epidemic forms of viral encephalitis most often follow an arbovirus infection, carried by arthropods (mosquitoes and ticks) from some natural host (e.g., horses) to man. The arboviral encepha-litides include eastern equine, western equine, St. Louis, Japanese B, West Nile, and California encephalitis. Herpes simplex virus encephalitis is the most common type of sporadic viral encephalitis and the most common cause overall. The typical patient is a young and previously healthy adult who suddenly develops alteration of consciousness, followed rapidly by the onset of seizures and a focal neurologic deficit. Herpes simplex causes an acute, focal, necrotizing encephalitis with inflammation and edema; MRI may show abnormalities in the medial and inferior temporal lobe on the involved side.
Neurosyphilis is a specific subtype of tertiary syphilis and occurs after a long latent period following the primary and secondary stages. Although the manifestations are protean, several specific syndromes are recognized: tabes dorsalis, general paresis, and meningovascular syphilis. Early Lyme disease may present as meningitis, cranial nerve palsies, and radiculoneuritis. The late phase may consist of encephalopathy, encephalomyelitis, and polyradiculoneuropathies. In addition to predisposing to a variety of other infections or conditions, the HIV virus can directly infect the CNS, producing meningitis or encephalitis. With well-established disease, AIDS patients may develop a number of different neurologic syndromes, including cerebral toxoplasmosis, cytomegalovirus encephalitis, cryptococcal meningitis, tuberculous meningitis, neurosyphilis, CNS lymphoma, dementia, myelopathy or myelitis, polyradiculopathy, neuropathy, and inflammatory myopathy. Progressive multifocal leukoencephalopathy (PML) occurs frequently, as does CNS lymphoma. Other important but uncommon neurologic infections include Creutzfeldt-Jakob disease, subacute sclerosing panencephalitis, rabies, and poliomyelitis. Some infections (e.g., botulism, tetanus, and diphtheria) and infestations cause neurologic manifestations by the elaboration of a toxin that affects the nervous system.
Inflammatory and Autoimmune Disorders
Some disorders are characterized by a pathologic picture of inflammation but are not known to be infectious. Examples include neurosarcoidosis, Behcet’s disease, acute and chronic inflammatory demyelinating polyradiculoneuropathy, acute disseminated encephalomyelitis (ADEM), and transverse myelitis. Clinically obvious neurologic involvement occurs in 5% to 15% of patients with sarcoidosis. The most common neurologic manifestation of sarcoidosis is facial nerve palsy, which may be bilateral. Behcet’s disease is a disorder of obscure pathogenesis. The primary manifestations are recurrent oral or genital ulcerations, ocular disease, primarily uveitis, and involvement of multiple organ systems. The most common neurologic complication is recurrent meningoencephalitis. Behçet’s disease is primarily relevant in the differential diagnosis of MS. The nervous system may be secondarily involved in systemic autoimmune disorders such as systemic lupus erythematosus (SLE) and related conditions, or when the autoimmune process is directed against blood vessels.
Trauma
The clinical effects of trauma to the head involve complex dynamics since one moving body, the brain, is traveling in relation to another moving body, the skull. With abrupt deceleration, the most frequent injury mechanism, the brain, suspended inside the skull, can impact against the inner table and the rigid meningeal structures, causing coup and contrecoup injuries. The areas most frequently injured are the frontal and temporal tips and the subfrontal regions. Closed, or nonpenetrating, head injuries are those in which no fracture or only a simple linear fracture occurs, without displacement of the fragments, rupture of the dura, or penetration or exposure of the brain substance. Penetrating, or open, head injuries are those with either compound or depressed fractures or penetrating wounds. In diffuse axonal injury, there is widespread disruption of axons, which can produce very significant clinical deficits. The most common type of closed head injury is simple concussion, producing brief loss of consciousness with no focal abnormalities on examination and normal imaging studies. Severity of injury correlates best with duration of loss of consciousness and length of anterograde amnesia. The Glasgow Coma Scale is commonly used in the evaluation and management of patients with craniocerebral trauma (see Chapter 51). Head injuries with Glasgow Coma Scale scores of greater than 12 are considered mild; 9 to 12, moderate; and less than 9, severe. Cerebral contusion is a more severe form of closed head injury in which there is superficial hemorrhage over the cortex. The most severe closed head injuries may produce ICH. Complications of compound and depressed skull fractures and of severe contusions and lacerations include cerebral edema, intracerebral hematoma, epidural or subdural fluid collections, SAH, post-traumatic meningitis and brain abscess, focal cerebral cicatrices, osteomyelitis of the skull, traumatic pneumocephalus, arteriovenous aneurysm, organic brain syndrome, and posttraumatic epilepsy.
Subdural hematomas (SDH) arise from torn veins bleeding into the subdural space. Acute SDH most often follow obvious head trauma. The rapidly expanding intracranial mass produces impairment of consciousness and focal signs. Chronic SDH develop more slowly and often follow minor head trauma, particularly in elderly patients. The intracranial mass effect develops slowly and typically produces impairment of consciousness with only minor focality on examination. Epidural hematomas most commonly follow fractures through the thin temporal squamosa of the skull that lacerate the middle meningeal artery. Patients may have a lucid interval following the injury, only to lapse into coma as the hematoma expands minutes to hours later.
Pharmaceuticals and Other Chemical Agents
Neurologic complications of pharmaceuticals and similar agents, such as vitamins and supplements, are a common problem. These range from the relatively common and innocuous, such as dizziness due to drug-induced orthostatic hypotension, or headaches due to a prescription medication, to the catastrophic, such as PML due to natalizumab. Drugs may cause neurologic effects because of intoxication, either accidental or purposeful, withdrawal or because of adverse reactions. Patients may become habituated or addicted to properly prescribed and administered drugs, as in opiates for chronic pain or sedative-hypnotics for sleep, so distinguishing between substance use and abuse becomes murky. This section deals with prescription medications, vitamins, and substances. Substance abuse is considered below.
Drugs may have similar efficacy and a similar side effect profile, termed class effects. The manifestations of intoxication related to drugs of a given class are referred to as toxi dromes (Table 53.10). Drug effects and side effects may mimic naturally occurring neurologic disease. The possibility of drug intoxication or withdrawal should be raised in any patient with AMS, delirium, or confusion.
TABLE 53.10 Toxidromes and Associated Drugs
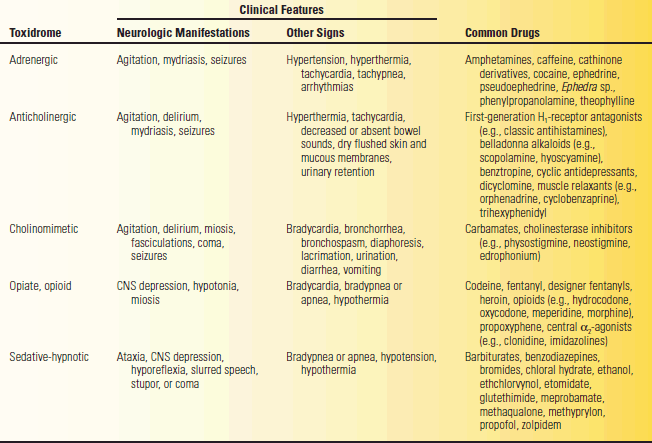
Modified from Ford, MD. Acute poisoning. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia: Elsevier/Saunders, 2012.








