Diseases of the Motor Unit
Harvey B. Sarnat
John H. Menkes
But this form of myopathy, this pseudo-hypertrophic paralysis which was described by Duchenne (of Boulogne), that great worker in neuro-nosography, is so different in its clinical characters from the progressive spinal amyotrophies that they have rarely been confused clinically. Pseudo-hypertrophic paralysis is a disease of early youth. It is scarcely ever met with after twenty years of age. It is noticed that the child becomes clumsy in his walk, that he is more easily fatigued than the other children of his age; for it is always, quoting from Duchenne’s description, in the lower extremities where it commences. Then the upper extremities may be attacked in their turn; but, whatever be the degree of the affection, the hands are generally absolved. Finally the muscles attacked, or at least a great number of them, present an augmentation of volume, an enormous increase in size, giving to the limb, or a segment of the limb, Herculean proportions. Anatomically this hypertrophy is characterized by lesions of the interstitial tissue, such as does not exist in the same degree in spinal amyotrophies. Moreover, and this is a peculiarity which is not found in Duchenne-Aran disease, heredity plays a great part in the development of pseudo-hypertrophic paralysis of the muscles. It often happens that several children are attacked in one family, and that some of their relatives may present the same affection.
—J. M. Charcot, Clinical Lectures on Diseases of the Nervous System, 1889.
The motor unit consists of the motor neuron, its axon, the neuromuscular junction, and muscle fibers that this single motor neuron innervates. The size of the motor unit varies greatly: Finely tuned small muscles, such as the extraocular muscles or the stapedius, have a 1:1 ratio, whereas large muscles not requiring such refined control, such as the glutei or quadriceps femoris, have a ratio of 1:200 or more. This chapter discusses disorders of the motor unit, or neuromuscular disorders, and excludes suprasegmental disorders of cerebral origin, such as spasticity, ataxia, and dyskinesias, that are not primary disturbances of the motor unit even though they may profoundly affect muscular function.
Neuromuscular disorders have a limited clinical expression with considerable overlap of symptoms and signs; hence, their definitive diagnosis depends in part on the proper application of laboratory techniques. The procedures used most are serum myogenic enzymes, imaging of muscle, electromyography (EMG), electrophysiologic measurement of motor and sensory nerve conduction velocities, and muscle biopsy. The latter procedure involves histochemistry, immunocytochemistry, biochemistry, and electron microscopy. Advances in genetics now provide molecular genetic markers for many specific diseases that may be diagnosed from a blood sample. Examples include many muscular dystrophies, spinal muscular atrophy (SMA), and several metabolic myopathies. The clinician must decide which of these procedures provides the needed information in the least invasive manner. At times, two procedures are complementary rather than competitive: Nerve conduction velocity and nerve biopsy provide different information. At other times, one procedure confirms another: A blood polymerase chain reaction for Duchenne dystrophy is nondiagnostic in approximately one-third of cases, but the more precise dystrophin immunoreactivity of the muscle biopsy is diagnostic in all cases. Several excellent texts and review chapters deal with disorders of muscle: Engel and Franzini-Armstrong (1), Brooke (2), Dubowitz (3), Swash (4), and Sarnat (5). The best current monograph on pediatric disorders of peripheral nerve is by Ouvrier and colleagues (6).
LABORATORY INVESTIGATION OF NEUROMUSCULAR DISEASE
Muscle Enzymes
Degenerating or necrotic myofibers release several enzymes into the serum because of loss of integrity of the sarcolemmal membrane. These provide useful markers in blood for a few neuromuscular diseases, but not for those
in which no degenerative changes occur or the process is so mild that renal clearance can compensate.
in which no degenerative changes occur or the process is so mild that renal clearance can compensate.
The gold standard of muscle enzymes is creatine kinase (CK), previously known as creatine phosphokinase. This enzyme of striated muscle is shared by only two other organs— the cardiac muscle and central nervous system (CNS)—and not by the liver, pancreas, or other viscera. If elevated, isoenzymes may be measured in blood to separate the source: The MM band is striated muscle, MB band is heart, and BB is brain or spinal cord. The upper range of normal serum CK in childhood is approximately 180 IU/L, and the CK usually must be approximately three times normal to be considered significant. Strenuous physical activity always results in a transient increase in serum CK, as shown in studies of athletes, so the enzyme should be measured after a period of relative rest. It also is elevated after traumatic injury of muscle; hence, it should not be measured for at least a week after muscle biopsy or EMG. It is transiently elevated in neonates because of minor contusions of muscle during labor and delivery, but is not elevated in infants delivered by cesarean section unless they have suffered prolonged labor and arrested descent within the maternal pelvis.
The serum CK may be mildly elevated (in the hundreds) in some neuromuscular disorders (e.g., SMA, limb-girdle muscular dystrophy), greatly elevated (in the thousands) in others (e.g., Duchenne and Becker muscular dystrophy, rhabdomyolysis), and remains normal in most (e.g., the congenital myopathies, mitochondrial cytopathies). In some myopathies, it is variable from normal to high (e.g., polymyositis, dermatomyositis), depending on the activity of the inflammation and necrosis. The serum CK is, therefore, not a screening test for neuromuscular diseases, but a normal CK level always excludes Duchenne muscular dystrophy, even in the presymptomatic neonate.
Other muscle enzymes are not as specific as CK and may be misleading. The first such serum enzyme discovered was aldolase, but this enzyme is expressed in many tissues, including in red blood cells where it is present in high concentrations. An elevated aldolase out of proportion to CK in the same sample is usually an artifact caused by hemolysis. The transaminases (e.g., aspartate aminotransferase, alanine aminotransferase), often regarded as liver enzymes, also are present in muscle, and some children with myopathy have undergone unnecessary liver biopsy for unexplained high liver enzymes because CK was not also measured. Enzymes other than CK rarely contribute to the diagnosis if CK is available.
Imaging of Muscle
In recent years, ultrasonography, computed tomography, and magnetic resonance imaging (MRI) have been applied to muscle in various neuromuscular diseases and indeed may be helpful at times in diagnosing fatty or fibrous connective tissue replacement of muscle and distinguishing edematous or mostly dystrophic myofibers from normal muscle. The most useful of these methods is ultrasound. It is probably the simplest, least invasive, and least expensive (7,8). Plain roentgenography of the extremities may provide useful information about the ratio of the muscle cylinder and subcutaneous fat pad and about intramuscular calcifications, but ultrasonography should be reserved for soft tissues, not for bone as in the usual radiographs of the extremities.
Cardiac Evaluation
In some myopathies, cardiomyopathy may be present as well as involvement of striated muscle (e.g., Duchenne muscular dystrophy, Emery-Dreifuss muscular dystrophy, some cases of nemaline rod myopathy), or there may be a disturbance in the Purkinje conduction system (e.g., myotonic muscular dystrophy). Electrocardiography, roentgenography of the chest, and echocardiography may be indicated in selected cases. In muscular dystrophies in which cardiac involvement to some degree is almost universal at some stage in the disease, early consultation with a pediatric cardiologist should be requested, even if the patient is asymptomatic, as a baseline for anticipated future problems.
Molecular Genetic Markers
Several molecular genetic DNA markers in blood now provide definitive diagnoses in specific diseases such as SMA and some cases of Duchenne dystrophy, and may obviate the need for more invasive diagnostic tests such as muscle biopsy. Muscle dystrophin is characterized and quantitated by means of the Western blot test or quantitated by immunohistochemistry or enzyme-linked immunosorbent assay (ELISA) (9). The polymerase chain reaction (PCR) blood test for Duchenne dystrophy is falsely normal, however, in approximately one-third of cases, but the more specific immunocytochemistry of antibodies against various portions of the dystrophin molecule is diagnostic in all cases and must be performed on frozen sections of muscle biopsy tissue. The ratio of myofibers expressing or not expressing dystrophin shown by specific antibodies to the rod domain, N-terminus, and C-terminus of the dystrophin molecule distinguishes Duchenne from Becker dystrophies and also detects the female carrier state and mildly affected girls. Immunocytochemical markers in the muscle biopsy for merosin (congenital muscular dystrophy), the family of sarcoglycans (limb-girdle muscular dystrophy), dystroglycans, collagen VI, dysferlin, caveolin-3 and others are now available as well. Genetic markers for many other diseases undoubtedly will become commercially available in the next few years. These markers have an additional importance in prenatal diagnosis and for the detection of carrier states.
Electrophysiologic Studies
Motor and sensory nerve conduction velocities (NCV) are valuable diagnostic studies of children with neuropathies. They also help to distinguish motor neuron disease from peripheral neuropathy and axonal degeneration from primary demyelination. NCV can be measured in the facial nerve and the phrenic nerve as well as those of the extremities. NCV usually can be performed with surface electrodes on the skin, without the use of needles, and hence are less frightening to the child and are not painful.
EMG requires needle insertion into muscle and is less useful in pediatric conditions than in those of adults. EMG may provide useful information in distinguishing myopathy from neuropathy, but does not generally distinguish the exact type of myopathy; therefore, it is not an alternative to the more specific muscle biopsy in most cases. EMG has the advantage of being able to sample several muscles at one examination, to compare distal and proximal muscles, upper and lower extremities and axial muscles, and any presence of left and right asymmetries. In myopathies, the EMG characteristically shows a shortened mean duration and lower amplitude of the motor unit action potentials. A unique pattern is found in myotonic conditions. In contrast to myopathies, neuropathic patterns are characterized by spontaneous activity at rest and motor unit action potentials of increased duration and amplitude.
EMG with repetitive nerve stimulation (i.e., evoked potential EMG) is a highly specific diagnostic test for myasthenia and is an example of one condition in which EMG is more definitive than the muscle biopsy. Other specialized EMG studies such as macro-EMG and single-fiber EMG recording are available in some centers but are not generally used for clinical diagnosis.
Muscle Biopsy
A muscle biopsy has become the most essential diagnostic procedure in evaluating a child with neuromuscular disease. It is a simple procedure generally performed in day-surgery under local anesthesia with a regional nerve block and does not require a general anesthetic. Needle biopsy has become popular in some centers. It can be performed percutaneously and has the advantage that repeated biopsies may be performed, if necessary (10). However, an open biopsy usually provides a better quality specimen for pathology and any biochemical studies.
The first decision is to choose the muscle to undergo biopsy. The selected muscle should be affected clinically, but not to such an advanced degree that all muscle tissue has degenerated beyond recognition. Additionally, the muscle should not have been subjected to EMG study in the previous 4 to 6 weeks. Because all muscles do not normally have the same ratio of fiber types, this is an additional consideration. The deltoid, for example, normally expresses a 60% to 80% predominance of type I fibers, which at times can complicate interpretation of neurogenic changes. The muscle most studied and generally suitable for the diagnosis of most generalized neuromuscular diseases is the vastus lateralis (i.e., quadriceps femoris), a large proximal muscle not in the region of large blood vessels or major nerves.
The muscle sample is divided, and portions are used for routine examination, histochemistry, electron microscopy, and biochemical or enzymatic studies. For routine histologic examination, a portion of the specimen can be fixed in 10% buffered formalin and embedded in paraffin. For histochemical and most histologic studies, the tissue is frozen in liquid nitrogen and then stored at -70°C. A separate piece of the biopsy should be frozen directly and preserved for possible biochemical studies.
The histochemical reactions generally used, together with their indications, are shown in Table 16.1. Histochemical studies have shown that muscle fibers in human skeletal muscle can be differentiated into two types: type I fibers, which are rich in oxidative enzymes but poorer in glycogen and phosphorylase, and type 2 fibers, weaker in oxidative enzymatic activities but rich in glycogen and phosphorylase. Calcium-mediated myofibrillar adenosine triphosphatase (ATPase) is equally strong in both fiber types, but epitopes are selectively inhibited by changing the pH of preincubation, so that at alkaline pH (10.4 or 9.8) type 2 fibers appear darker than type 1, and at acid pH (4.7 and 4.3) type 1 fibers are darker, with subtypes of 2 appearing as intermediate intensity of staining and thus providing a
convenient marker of these subtypes. Type 2C fibers are the least mature type and represent about 5% of fibers in term neonates, more in preterm infants and are infrequent in adult muscle. Normal muscle of children and adults presents a mosaic of both types 1 and 2 fibers. A fuller discussion of histochemical techniques, the histochemistry of developing muscles, and the histologic changes in neurogenic muscular atrophies is presented by Dubowitz (3), Sarnat (5), and Engel and Franzini-Armstrong (1).
convenient marker of these subtypes. Type 2C fibers are the least mature type and represent about 5% of fibers in term neonates, more in preterm infants and are infrequent in adult muscle. Normal muscle of children and adults presents a mosaic of both types 1 and 2 fibers. A fuller discussion of histochemical techniques, the histochemistry of developing muscles, and the histologic changes in neurogenic muscular atrophies is presented by Dubowitz (3), Sarnat (5), and Engel and Franzini-Armstrong (1).
TABLE 16.1 Histochemical Reactions for Muscle Biopsies | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Immunocytochemistry has become important in muscle biopsy diagnosis only in the past few years. Frozen sections are required for sarcolemmal region proteins, such as dystrophin, spectrin, merosin, and the sarcoglycans (11). Intermediate filament proteins such as vimentin and desmin are important in some congenital myopathies and can be demonstrated both in frozen and paraffin sections. Heavy-chain myosin immunocytochemistry is the first technique to reliably distinguish fiber types in paraffin sections. Other special purpose immunocytochemical methods include antibodies against actin, tropomyosin, and for marking T and B lymphocytes and macrophages to help distinguish different types of inflammatory myopathies.
Electron microscopy is needed only in selected cases, particularly when there is a question of a mitochondrial myopathy (see Chapter 2). However, tissue submitted for electron microscopy can be used to prepare 1-μ epoxy resin-embedded sections that are suitable for examination in the light microscope after staining with toluidine blue. These thin sections are valuable to confirm the presence of inclusions, such as those seen in a variety of storage diseases, and other details. Thinner sections for electron microscopy usually are stained with uranyl acetate and lead citrate.
Biochemical Studies
In metabolic myopathies, particularly in the glycogenoses and lipid storage myopathies, quantitative assays may be performed on frozen muscle biopsy tissue for specific enzymatic activities, such as acid maltase, myophosphorylase, brancher and debrancher enzymes, phosphofructokinase, and carnitine palmitoyl-transferase. Total muscle glycogen and fat and muscle carnitine content also may be measured. These tests are expensive and not routine; they are requested only if the clinical suspicion is strong and especially if the muscle biopsy histochemistry and ultrastructure support the diagnosis.
The increased awareness of mitochondrial diseases and the large number of different deletions and point mutations involving mtDNA have resulted in a much larger number of muscle biopsies now being performed with the specific question of ruling out a mitochondrial cytopathy. Quantitative analysis of each of the mitochondrial respiratory chain enzymes and of mtDNA can be performed on freshly frozen muscle biopsy tissue if there is sufficiently strong clinical or histochemical and ultrastructural evidence. A few mitochondrial cytopathies fail to exhibit morphologic or histochemical changes, so normal routine biopsy results do not definitively exclude mitochondrial disease, but these cases are only a minority of the mitochondrial cytopathies. Frozen tissue remaining on the block after cutting frozen sections is not suitable for the quantitative analysis of mitochondrial respiratory complexes because the isopentane used to preserve the histologic detail and prevent ice crystal artifacts during the freezing process in liquid nitrogen interferes with the determination of complexes I through III and gives falsely low values. A portion of the muscle biopsy should be freshly frozen directly for possible later biochemical study (see Chapter 2).
DISEASES OF THE MOTOR NEURON
In children, the most common entity affecting the motor neuron in the brainstem and spinal cord is the SMAs. Poliomyelitis, once seen in epidemic proportions, is a rarity in Western countries. Other neurotropic agents including West Nile virus (12) and other enteroviruses that, like poliomyelitis, produce transient or permanent paralysis are considered in Chapter 7. A flaccid paralysis resembling poliomyelitis has been seen in some children who are recovering from an acute episode of asthma (Hopkins syndrome) (see Chapter 17). A familial degenerative disease resembling the adult form of amyotrophic lateral sclerosis (ALS) is encountered rarely in pediatric practice. It is characterized by degeneration of the pyramidal tracts and the motor cells of the spinal cord and brainstem (see Chapter 3). Involvement of the motor neurons as part of generalized storage diseases is observed in some hereditary metabolic diseases such as GM1 gangliosidosis, Hurler syndrome, infantile Gaucher disease, glycogenosis type II (Pompe), and neuronal ceroid-lipofuscinosis. These entities are considered under their appropriate sections in Chapter 1.
Spinal Muscular Atrophies
The SMAs are a group of relatively common diseases occurring in infancy or early childhood, transmitted by an autosomal recessive gene and manifested by widespread muscular denervation and atrophy (13). Carrier frequency has been estimated to fall between 1 in 50 to 80, and the disease has an overall incidence of 1 in 10,000 to 25,000, making it the second most common hereditary neuromuscular disease after Duchenne dystrophy. Three forms of SMA have been recognized. These are the infantile, acute form (SMA type 1 or Werdnig-Hoffmann disease), the intermediate form (SMA type 2), and the juvenile form (SMA type 3 or Kugelberg-Welander disease). Some workers also have delineated an adult form of SMA (SMA type 4), in which symptoms start after 30 years of age. A severe fetal form of SMA is described as type D. The muscle biopsy in
this entity may present a picture of centronuclear myopathy, thus confusing the diagnosis (13a,b). Approximately one-fourth of cases of SMA fall into the type 1 variety, approximately one-half into type 2, and the remainder into type 3 (2). All three forms are caused by mutations in a gene, the telomeric survival motor neuron gene (SMN1) (14). Two copies of the gene are normally present, a telomeric copy, SMN1, and a centromeric copy, SMN2. The two genes have greater than 99% homology, and their exons differ in but two nucleotides (15). SMN1 produces a majority of full-length SMN transcript, whereas SMN2 generates mostly a nonfunctional isoform lacking the carboxy-terminal amino acids encoded by exon 7.
this entity may present a picture of centronuclear myopathy, thus confusing the diagnosis (13a,b). Approximately one-fourth of cases of SMA fall into the type 1 variety, approximately one-half into type 2, and the remainder into type 3 (2). All three forms are caused by mutations in a gene, the telomeric survival motor neuron gene (SMN1) (14). Two copies of the gene are normally present, a telomeric copy, SMN1, and a centromeric copy, SMN2. The two genes have greater than 99% homology, and their exons differ in but two nucleotides (15). SMN1 produces a majority of full-length SMN transcript, whereas SMN2 generates mostly a nonfunctional isoform lacking the carboxy-terminal amino acids encoded by exon 7.
Pathologic Anatomy and Pathogenesis
Both Guido Werdnig, an Austrian neurologist from the University of Graz (16), and Johan Hoffmann, a German neurologist (17), in their original descriptions of the infantile form of SMA, pointed to the conspicuous loss of ventral horn cells along the entire length of the spinal cord. Some of the residual motor neurons are in the process of degenerating or are being phagocytized by microglial cells. Motor neurons in the brainstem, notably in the hypoglossal and motor trigeminal nuclei also are affected. Those cranial nerves innervating extraocular muscles and sacral motor nerves innervating the striated muscle of the urethral and rectal sphincters are selectively spared. Some authors describe suprasegmental lesions, including loss of large pyramidal cells from layers 5 and 6 of the motor cortex. These findings must be interpreted in light of the severe agonal anoxia that is commonly encountered. In general, the absence of upper motor neuron involvement is an important clinical feature that distinguishes SMAs from degenerative motor neuron diseases such as ALS.
More than 95% of patients experiencing the various forms of SMA have a homozygous deletion or absence of exons 7 and 8 for both copies of the SMN1 gene (13). Subjects who do not have detectable deletions have microdeletions or point mutations in the gene, or have their SMN1 gene converted to SMN2 (18,19). In some patients with SMA types 2 and 3, SMN1 is not deleted; rather, it is replaced by SMN2, the reverse form of “gene conversion” (19,20). Two other genes have been mapped to the SMA critical region. Homozygous deletions of neuronal apoptosis inhibitory protein gene (NAIP) are seen in up to 69% of patients with SMA type 1. By contrast, only some 12% to 18% of patients with SMA types 2 and 3 are deleted for this gene (21). A third gene in this region, shown to encode a subunit of the basal transcription factor (TFIIH) is deleted in approximately 15% of all types of SMA (21). NAIP and TFIIH as well as a multicopy microsatellite marker in close proximity to SMN1 have been proposed as SMA-modifying genes (22,23).
The genetic basis for the phenotypic variability of SMA is not completely clear. About one-half of severely affected SMA1 children also miss both homologues of a neighboring gene, the gene for neuronal apoptosis inhibitory protein (NAIP). Milder forms of SMA appear to have more than two copies of SMN2, and in asymptomatic or late-onset subjects with homozygous deletion of the SMN1 gene, there were four copies of the SMN2 gene (24,25).
The SMN protein is widely expressed in the cytoplasm and nucleus of brain, the motor neurons, and in a variety of non-CNS tissues (26). In the nucleoplasm the SMN protein is localized to discrete bodies called “gems” where it forms a stable complex with a group of proteins called the gamins (27). This complex is intimately involved in the assembly of spliceosomal small nuclear ribonucleoproteins, which function in the splicing of RNA. The reason for the effect of gene mutation on the spinal motor neurons is still unclear, and it is not known whether symptoms result from deficiencies in SMA functions that are specific to the motor neuron, or common to all cells but at a higher demand in the motor neuron. One attractive hypothesis is that SMN protein has an antiapoptotic effect (28), suggesting that that SMA is a primary disorder of apoptosis, with the condition representing a physiologic process that becomes pathologic because of continued degeneration of motor neurons in late fetal life as well as postnatally (29,30).
Prominent glial proliferation often occurs in the proximal portion of the anterior spinal roots, an abnormality initiated in fetal life, and which was thought to induce secondary neuronal degeneration (31). In some instances, the glial bundles also are demonstrated in the posterior roots, accompanied by shrinkage of the dorsal root ganglion cells (32). An inconstant finding in SMA is loss of myelin from the posterior columns of thoracic and lumbar segments and in the corticospinal tracts (33). These phenomena are now believed to be secondary reactive changes. Removal of the peripheral target of the anterior horn cells accentuates the normal process of motor neuron apoptosis (34,35,36), but there is no evidence that the primary disorder in this disease is in the muscle target or that there is a distal-to-proximal dying-back axonal degeneration in the nerve.
There is selective loss of the large motor neurons, but a striking preservation of several types of motor neurons, notably some of the cervical cord (37) and those innervating extraocular muscles. This histologic contrast is expressed clinically as preservation of normal eye movements, even in late stages of the disease. Some genes that program the differentiation and maintenance of motor neurons are expressed in all motor neurons, whereas others are expressed only in certain motor neurons and may provide a protective effect on oculomotor neurons by the principle of genetic redundancy (see Chapter 5 on neuroembryology).
Clinical Manifestations
The clinical picture of SMA is marked by reduction of muscle power and spontaneous movement (38). Muscle
weakness is symmetric and is more extensive in the proximal part of the limbs, and what little movement is left to the child is found in the small muscles of the hands and feet. At the same time, the affected muscles undergo atrophy, although this is concealed by the subcutaneous fat normally seen at this age. Muscles of the trunk, neck, and thorax are affected equally, but the diaphragm generally is spared until the late stages of the disease (39) (Fig. 16.1). Cardiac and smooth muscles are usually spared as well. With progression of the disease, involvement of bulbar musculature becomes more prominent, and atrophy and fasciculation of the tongue are noted. Deep tendon reflexes are nearly always markedly reduced or absent. There is no intellectual retardation or sphincter disturbance. Sensory nerve involvement has been demonstrated by nerve conduction studies and confirmed by demonstration of axonal loss on sural nerve biopsy and autopsy. Hypertrophy of unmyelinated axons of the sural nerve also is frequent (29). These sensory nerve changes have not been observed in SMA types 2 and 3 (40,41).
weakness is symmetric and is more extensive in the proximal part of the limbs, and what little movement is left to the child is found in the small muscles of the hands and feet. At the same time, the affected muscles undergo atrophy, although this is concealed by the subcutaneous fat normally seen at this age. Muscles of the trunk, neck, and thorax are affected equally, but the diaphragm generally is spared until the late stages of the disease (39) (Fig. 16.1). Cardiac and smooth muscles are usually spared as well. With progression of the disease, involvement of bulbar musculature becomes more prominent, and atrophy and fasciculation of the tongue are noted. Deep tendon reflexes are nearly always markedly reduced or absent. There is no intellectual retardation or sphincter disturbance. Sensory nerve involvement has been demonstrated by nerve conduction studies and confirmed by demonstration of axonal loss on sural nerve biopsy and autopsy. Hypertrophy of unmyelinated axons of the sural nerve also is frequent (29). These sensory nerve changes have not been observed in SMA types 2 and 3 (40,41).
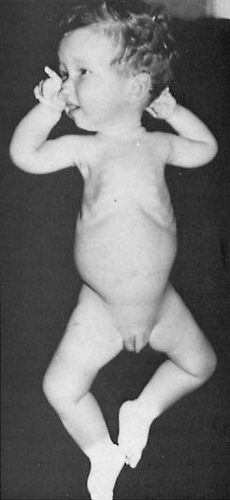 FIGURE 16.1. A 1-year-old infant with severe spinal muscular atrophy showing intercostal recession and diaphragmatic breathing. Note frog-leg posture, ability to flex elbows and move hands, and normal facial expression. (Courtesy of Professor Victor Dubowitz, Royal Postgraduate Medical School, London.) |
TABLE 16.2 Age of First Clinical Manifestations in Infantile Spinal Muscular Atrophy | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
The age at which the first clinical manifestations become apparent is presented in Table 16.2. It is evident from this table that there are at least two populations, one with onset of symptoms before 6 months of age, SMA type 1, and another with its onset after 6 months of age, SMA types 2 and 3. About 10% of infants with SMA type 1 are born with arthrogryposis, and this may indicate the severe fetal form recently designated type 0 (13a,b).
In SMA type 1 (infantile SMA, Werdnig-Hoffmann disease), the onset is acute, and the disease progresses rapidly. Infants who were already severely hypotonic at birth rarely survive the first year of life, whereas those whose weakness appears postnatally tend to deteriorate more slowly (42,43). Some infants can even experience transient improvement. This improvement is partly caused by a true stationary period in the disease, such as was already noted by Hoffmann in one of his earliest patients (17). Maturation of partially paralyzed muscles also can give the impression of improvement. In most instances, however, the disease is fatal by 3 years of age, with death generally resulting from a respiratory infection.
Children who experience SMA type 2 [29% in Brandt’s series (38), 47% in the more recent series (2)] develop normally for the first 6 months of life, but by 18 months of age experience an arrest of their motor milestones. These children generally lack bulbar symptoms, and their disease has a less malignant course (43). One unusual feature of this form of SMA is the presence of a tremor that affects the upper extremities (44) and can even be recorded on the electrocardiogram as a tremor of the baseline (45). Serum CK activity is elevated to approximately five times normal in approximately one-half of these patients but is never in the thousands of units as in muscular dystrophy; hypertrophy of the gastrocnemius is not unusual (46).
SMA type 3, a milder form of SMA consistent with survival into adult life, was first described by Wohlfart and coworkers (47) and by Kugelberg and Welander (48). Muscular weakness develops after 18 months of age, and in some patients does not manifest until adult life. Adult onset SMA is referred to as SMA type 4. It is characterized by involvement of the predominantly proximal muscles and
thus bears close clinical resemblance to the muscular dystrophies. Unlike SMA types 1 and 2, impaired joint mobility is relatively common. The condition is differentiated from Duchenne muscular dystrophy by a less striking elevation in muscle enzymes, and from the other types of muscular dystrophies by the alterations seen on muscle biopsy (see Muscle Biopsy, later in this chapter). In some families, SMA types 1 and 2, or types 2 and 3, coexist. This observation represents a genetic puzzle because haplotype analysis has shown that this variability cannot be accounted for by different alleles at the SMA locus (49).
thus bears close clinical resemblance to the muscular dystrophies. Unlike SMA types 1 and 2, impaired joint mobility is relatively common. The condition is differentiated from Duchenne muscular dystrophy by a less striking elevation in muscle enzymes, and from the other types of muscular dystrophies by the alterations seen on muscle biopsy (see Muscle Biopsy, later in this chapter). In some families, SMA types 1 and 2, or types 2 and 3, coexist. This observation represents a genetic puzzle because haplotype analysis has shown that this variability cannot be accounted for by different alleles at the SMA locus (49).
In some families, a condition that clinically resembles Kugelberg-Welander disease is transmitted in an autosomal dominant manner (50). X-linked pedigrees also have been reported. Hexosaminidase A deficiency can resemble Kugelberg-Welander disease, but patients demonstrate mental deterioration (51) (see Chapter 1).
A condition in which spinal muscular atrophy presents with early respiratory distress, and in which there also is intrauterine growth retardation, foot deformities, early involvement of the diaphragm, and a predominance of distal muscular weakness can be distinguished from SMA type 1. The gene for this condition has been mapped to chromosome 11 q13–q21. It encodes the immunoglobulin binding protein 2 (52).
Diagnosis
Genetic Studies
The definitive diagnosis may now be made by the marker in blood of the SMN gene that usually shows deletions of exons 7 and 8 (18). Thus, in the series of van der Steege and colleagues, the SMN1 gene, as determined by PCR, was homozygously absent or interrupted in 98.6% of SMA patients (53). An additional gene mapped to 11q13–q21 in SMA may help explain early respiratory failure in some patients (53a). Muscle biopsy, which until only recently had been the diagnostic standard, can therefore be avoided in most patients, with exception of the small fraction whose molecular genetic test results are negative or uninformative.
Biochemical Studies
The study of serum enzymes is important in the differential diagnosis of motor neuron disease. Serum CK is often normal in SMA type 1 but may be elevated in the hundreds, although never in the thousands. It usually is normal or only slightly elevated in the other types. The excretion of amino acids and creatine is generally normal during the early stages of the illness. None of these features is definitively diagnostic of SMA.
Neurophysiologic Studies
EMG findings help to confirm the clinical diagnosis of motor neuron disease. Characteristically, the denervated muscles contract spontaneously, involving single muscle fibers (fibrillation) or entire motor units (fasciculation) (54). The finding most specific for the disease and not observed in any other condition marked by muscular denervation is the presence of spontaneous, rhythmic muscle activity at a frequency of 5 to 15 per second, which can be activated by voluntary effort. This abnormality can be recorded in approximately 75% of patients, irrespective of age or severity of disease (55). In addition, the residual motor unit potentials are polyphasic and increased in amplitude and duration. With increased muscular effort, little increase occurs in the frequency of discharge and recruitment is impaired. Conduction velocity in the motor nerves is decreased in the more severely affected children (56). This is because of the preferential involvement of the largest motor neurons, which also have the most myelinated and fastest-conducting axons.
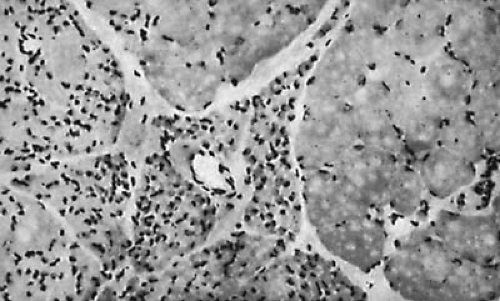 FIGURE 16.2. Transverse section of muscle from a patient with Werdnig-Hoffmann disease. Note the relative preservation of muscle fibers in the right side of the field and atrophy of fibers on the left (hematoxylin and eosin, × 20). (Courtesy of Dr. P. Cancilla, Department of Pathology, University of California, Los Angeles, UCLA School of Medicine.) |
Muscle Biopsy
Chemical analyses for glycogen, lipids, glycolytic enzymes, and mitochondrial enzymes are performed, with the selection of assays dictated by the clinical presentation.
In SMAs, a biopsy shows the classic features of denervation atrophy (Fig. 16.2), but perinatal denervation shows unique features not seen in neurogenic atrophy of mature muscle. Large patches of small, atrophic fibers of both histochemical types are present, and both scattered and grouped muscle fibers of exclusively type I also are present. The normal mosaic or chessboard pattern is lost. This uniformity of fiber type reflects an ongoing compensatory reinnervation (3). Electron microscopy adds little additional diagnostic information except to demonstrate scattered immature myofibers including myotubes (29, 54a). The myofilaments are loosened, and mitochondria are diminished in number and show atrophic cristae. Muscle biopsies showing the perinatal pattern of denervation-reinnervation nearly always signify SMA but are not definitively diagnostic because rare perinatal polyneuropathies may yield a similar picture.
TABLE 16.3 Diagnosis of 107 Cases of Floppy Infanta | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Differential Diagnosis
The presenting signs of the infant with SMA type 1 (Werdnig-Hoffmann disease) are poor muscle tone and a marked delay in motor development. Because a variety of well-defined diseases present the picture of the floppy infant (Table 16.3), differential diagnosis on purely clinical grounds can be difficult. Unlike Walton writing in 1957 (57), we have found the most common cause for diminished muscle tone in a small infant to be atonic cerebral palsy, often secondary to cerebellar hypoplasia. Hypotonia also may result from major abnormalities of the cerebrum, both developmental malformations and disorders acquired in the perinatal period. Invariably, affected infants have considerable intellectual retardation. Spontaneous movements are present, and the tendon reflexes are easily elicited. Often when the infant is lifted by the trunk, the legs promptly become rigid, and a striking accentuation of the extensor thrust reflex is seen. Although some of these children remain hypotonic, others develop dyskinesias or a clear-cut hypertonia within 1 to 3 years. A more extensive discussion of this condition is found in Chapter 6.
Amyotonia congenita, now an obsolete term, was a clinical syndrome reported briefly in a single case by Oppenheim in 1900 (58). It represents a number of unrelated disease processes. It is important to differentiate these entities, now collectively termed congenital myopathies (benign congenital hypotonias), from the usually fatal SMA type 1. The term congenital myopathy is used to describe floppy infants with a marked delay in motor development. Some of these children recover completely, whereas others can be considered to have a stationary or, at worst, a slowly progressive muscular disorder. In these infants, the muscles are soft and flabby, and a remarkable range of passive movement is possible. Deep tendon reflexes are elicitable, and spontaneous movements are more prominent than in SMA type 1. Respiratory muscles are involved only slightly, and intellectual development is usually normal. Histochemical and electron microscopic studies of muscle biopsies have delineated numerous distinct entities, many of which demonstrate specific genetic mutations (see Congenital Myopathies, later in this chapter).
A relatively common clinical problem is that of a floppy infant with delayed gross motor milestones, notably delayed walking, but with normal or nearly normal fine motor development and speech and no demonstrable muscular disorder. This heterogeneous entity, termed congenital laxity of ligaments by the late Dr. Frank Ford (59), has more recently been designated as dissociated motor development (60) or benign maturation delay (61).
In a series of such infants reported by Lundberg, 52% achieved normal development between 17 months and 6 years of age (60). A further 24% continued to have delayed motor milestones without any apparent cause. Ultimately, 15% were found to have mild mental retardation. The remaining infants experienced various neuromuscular disorders, notably congenital deafness with hypoactive labyrinths and macrocephaly (61,62). Muscle biopsy revealed fiber size disproportion and showed a predominance of type 1 fibers, which persisted despite clinical improvement (61). Dissociated motor development should be distinguished from the less common Ehlers-Danlos syndromes, which are characterized by marked hyperelasticity of skin.
The syndrome of congenital fiber type disproportion (Table 16.4) (63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84) is part of a broader spectrum of muscle dysmaturation. Type 1 fibers are uniformly small and also more numerous than type 2 fibers that often exhibit compensatory hypertrophy. Many infants who demonstrate dysmaturation of muscle, particularly when type 2 fibers are affected, also have significant developmental retardation (85).
Rarer causes for infantile hypotonia include various forms of congenital or neonatal myasthenia gravis, some of which can be diagnosed by the striking improvement in muscle strength after administration of an anticholinesterase drug. For diagnostic purposes, a slowly metabolized drug, such as neostigmine administered subcutaneously or intramusculary, but never intravenously, is more suitable for infants than edrophonium chloride, whose effectiveness is often too fleeting to allow correct sequential assessment of muscle strength. Muscular dystrophy is only rarely seen in a small infant, but in no other condition is a significant elevation in serum enzymes, particularly in CK, common. In the absence of a family history of a dystrophic process, the diagnosis depends mainly on a muscle biopsy.
Finally, infantile botulism (see Chapter 10), Down syndrome, transection of the spinal cord, congenital polyneuritis, muscular hypotonia owing to Marfan syndrome or Prader-Willi syndrome, various chronic illnesses, malnutrition, or metabolic disorders (e.g., organic acidurias and mitochondrial disorders) also must be considered in the differential diagnosis of the hypotonic child.
TABLE 16.4 Some Congenital Myopathies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Several rare conditions are transmitted as autosomal recessive traits in which amyotrophy of the limbs and of the bulbar musculature are combined with progressive pyramidal tract symptoms (juvenile ALS). These conditions are reviewed in Chapter 3.
Treatment
Treatment for SMA is purely symptomatic and is of no value in altering the course of infantile SMA type 1. In the forms with slower progression, treatment should be directed at maintaining joint mobility and avoiding contractures. The child should be fitted with a lightweight support for the spine to prevent kyphoscoliosis. If at all possible, the youngster should be encouraged to walk with braces. Active exercise can help strengthen still functioning muscles (2,45). Dysphagia may be a late complication in some patients, and aspiration is a risk.
Several workers have suggested that SMN2 gene expression can be increased by administration of 4-phenylbutyrate, an inhibitor of histone deacetylase, an enzyme that promotes transcriptional repression (86). This compound has been proposed for the treatment of SMA 1.
Arthrogryposis
Arthrogryposis refers to a syndrome of multiple congenital and nonprogressive contractures of the joints that are fixed in flexion or, less commonly, in extension, accompanied by diminution and wasting of skeletal muscle. The term is derived from the Greek “hooked joint.” The condition can be seen in isolation or can be accompanied by a variety of other congenital malformations, particularly clubfoot and cerebral maldevelopment (87,88).
Arthrogryposis appears to have several causes, all involving impaired fetal mobility (89). These can be categorized as myopathic, neuropathic, connective tissue disorders, and exogenous effects. Hall has classified arthrogryposis into conditions where there is primarily limb involvement, those in which there is limb involvement plus other body areas, and those in which there is limb and CNS involvement (88). In one group of infants (25% of an English series) (90), the neuromuscular apparatus was normal, and arthrogryposis was caused by external mechanical factors. Such factors include a malformed uterus (e.g., bicornuate uterus), the treatment of maternal tetanus with muscle relaxants, or oligohydramnios (e.g., Potter syndrome). In the majority of infants, arthrogryposis results from a neuromuscular or a combined cerebral and neuromuscular disorder (89,90). The most common of these disorders is one in which the large anterior horn cells are markedly reduced, the muscle fibers are small and often hyalinized, and the EMG abnormalities are consistent with denervation (91). In contrast to the SMAs, no gliosis occurs, and the small neurons in the ventral horn of the spinal cord are preserved or even increased in number (92). In the experience of Quinn and colleagues, neurogenic arthrogryposis constituted 30% of cases (93). A neurogenic arthrogryposis with velopharyngeal incompetence is associated with a deletion at 22q11.2 (93a).
In other cases of arthrogryposis, the changes are compatible with congenital muscular dystrophy (94), congenital myotonic dystrophy (95), congenital myasthenic syndrome (96), fibrosis of the anterior spinal roots, or evidence of embryonic denervation and maturation arrest of muscle (95). Such cases constituted 60% of subjects with arthrogryposis who came to autopsy (93).
The prevalence of the condition varies considerably, ranging from 1:3000 in Canada to 1:12,000 in Western Australia to 1:56,000 in Edinburgh (87,97). Even though autosomal dominant, autosomal recessive, and X-linked transmission have been well documented, a viral or toxic etiology is likely in the majority of cases. In at least one instance, maternal antibodies against the fetal form of the acetylcholine receptor (AChR) were demonstrated, and plasma from anti-AChR antibody–positive mothers who had borne offspring with severe arthrogryposis induced fixed joints in neonatal mice when injected into pregnant dams (98).
Arthrogryposis is marked by multiple and severe flexion and extension contractures of all extremities, notably the hips, elbows, and fingers. Skin creases are absent, an indication of early intrauterine onset (87), and approximately one-fourth of affected infants have contractures of the temporomandibular joint. These contractures produce the typical round face and micrognathia (99). In one variant, amyoplasia, the shoulders are internally rotated, the elbows are extended, the wrists are flexed, and the hands are clenched. In the lower extremities, the knees are generally flexed, and the feet are maintained in an equinovarus position. Some 10% to 30% of patients have various superimposed developmental malformations of the CNS (89).
In Pena-Shokeir syndrome I, a heterogeneous condition, arthrogryposis is accompanied by facial anomalies, pulmonary hypoplasia, intrauterine growth retardation, and a variety of cerebral malformations. There is a paucity of motor neurons and muscular atrophy (100). Pena-Shokeir syndrome type II manifests by arthrogryposis not associated with deficiency of motor neurons but is accompanied by microcephaly, ocular defects, and a variety of skeletal abnormalities (101). Several other malformation syndromes, some with chromosomal disorders, can be accompanied by arthrogryposis. These are listed in review articles by Hageman and coworkers (89), O’Flaherty (87), and Hall (102) and in the text by Jones (103).
Hall and coworkers (102,104) and Staheli and coworkers (105) distinguish several conditions termed distal arthrogryposis, which, in contrast to the proximal arthrogryposes, tend to be genetically transmitted and are marked by congenital contractures of hands and feet. The
hand of such infants is characteristically tightly fisted with tight adduction of the thumb and medial overlapping of the fingers, similar to the position of the hand in the trisomy 18 syndrome (see Chapter 4). In some families, distal arthrogryposis is dominantly inherited and appears sporadically in others. A condition termed distal arthrogryposis type II also can have involvement of the proximal limb joints and associated congenital anomalies. In a significant proportion of families, arthrogryposis type II is dominantly inherited. One of the more common dominantly inherited conditions is the Freeman-Sheldon (“whistling facies”) syndrome, in which arthrogryposis and a characteristic “whistling facies” is accompanied by microcephaly, mental retardation, and seizures (105,106). Generally, patients with distal arthrogryposis have a better prognosis in terms of limb function than those with mainly proximal joint involvement.
hand of such infants is characteristically tightly fisted with tight adduction of the thumb and medial overlapping of the fingers, similar to the position of the hand in the trisomy 18 syndrome (see Chapter 4). In some families, distal arthrogryposis is dominantly inherited and appears sporadically in others. A condition termed distal arthrogryposis type II also can have involvement of the proximal limb joints and associated congenital anomalies. In a significant proportion of families, arthrogryposis type II is dominantly inherited. One of the more common dominantly inherited conditions is the Freeman-Sheldon (“whistling facies”) syndrome, in which arthrogryposis and a characteristic “whistling facies” is accompanied by microcephaly, mental retardation, and seizures (105,106). Generally, patients with distal arthrogryposis have a better prognosis in terms of limb function than those with mainly proximal joint involvement.
The reduced fetal movements of arthrogryposis permit an in utero diagnosis by ultrasonography and, if necessary, by fetoscopy (107).
Treatment of arthrogryposis should commence immediately after birth, with passive motion exercises, braces, and casts (87). Subsequently, surgery is usually necessary, particularly for contractions of the lower extremities and hips. These procedures are contraindicated in the presence of considerable weakness and amyoplasia. Generally, functional improvement of extension contractures is better than that of flexion contractures, and arthrogryposis secondary to maternal factors has a better outlook than that owing to neuromuscular disorders (102,108). In selected cases, a muscle biopsy may be indicated to rule out myopathies, such as congenital muscular dystrophy, often associated with multiple congenital contractures.
DISEASES OF THE AXON
Disorders of the axon result from genetic, infectious, postinfectious, traumatic, or toxic processes, and are, therefore, best discussed under their respective headings (see Chapters 3, 7, 8, 9, and 10, respectively).
In the differential diagnosis of muscular weakness, a polyneuritic process should always be considered. Although, supposedly, the presence of sensory loss in the affected area would easily distinguish polyneuritis from other disorders of the motor unit, an isolated motor neuropathy is far from rare. Commonly, motor weakness can overshadow sensory disturbances, the latter being particularly difficult to demonstrate in infants.
In general, the polyneuritides have a rapid onset, a feature they share only with the inflammatory muscular disorders: polymyositis and dermatomyositis. The site of muscular involvement is often a clue to the cause of the weakness; whereas the SMAs and the muscular dystrophies involve the proximal musculature preferentially, the polyneuritides usually affect the distal musculature. Ascending paralysis such as was originally described by Landry, Guillain, and Barré (see Chapter 8) is uncommon in infants. As a result of the predilection of the polyneuritides for the distal musculature, ankle reflexes are lost early in the illness; in the muscular dystrophies, they are retained.
The distinction between infectious polyneuritis and the hereditary motor and sensory neuropathies rests on the slower evolution of the neuropathies, their frequent association with skeletal deformities or cerebellar involvement, and the presence of a similar neuropathic condition in other members of the immediate family. In approximately two-thirds of children with infectious polyneuritis, the spinal fluid is abnormal. A classic picture of albuminocytologic dissociation, namely an elevated protein level in the absence of a cellular response, is common during the first 2 to 8 weeks of the illness. Motor nerve conduction times are usually slowed, although they can be normal during the initial phases of infectious polyneuritis. Although slowed motor conduction time, when present, is characteristic of infectious polyneuritis, it also can be seen with severe atrophy in anterior horn cell disease (poliomyelitis, West Nile encephalitis, and SMA type 1). In approximately one-half of the patients, the sensory nerve conduction times also are delayed. The histologic changes in affected muscle are rarely significant during the acute phase of infectious polyneuritis, whereas in chronic polyneuritis, the main abnormality is a widespread muscular atrophy.
DISEASES OF THE NEUROMUSCULAR JUNCTION
Myasthenia Gravis
Myasthenia gravis, a chronic disease characterized by unusual fatigability of voluntary muscles, was first described by Willis in 1672 (109). Three forms of myasthenia gravis are seen in childhood: juvenile myasthenia gravis, congenital myasthenia gravis, and transient neonatal myasthenia gravis. Aside from age of onset, there is no difference in terms of pathology and pathogenesis between juvenile myasthenia gravis and adult-onset myasthenia gravis.
Pathologic Anatomy
Microscopic changes in myasthenia gravis can be minute, even in severely affected muscles. On electron microscopy, the nerve terminals are small, and the cleft between the nerve and the subneural apparatus is widened. Additionally, the area reactive for acetylcholinesterase is reduced, but with no abnormality in the number or size of synaptic vesicles. In 70% to 80% of patients, there are pathologic changes in the thymus. Usually, these consist of lymphoid
hyperplasia. Although thymomas occur in approximately 10% of all patients with myasthenia, they are rare in childhood. Myocardial abnormalities have been found in approximately half of autopsied patients.
hyperplasia. Although thymomas occur in approximately 10% of all patients with myasthenia, they are rare in childhood. Myocardial abnormalities have been found in approximately half of autopsied patients.
Pathogenesis
The functional lesion in juvenile myasthenia gravis is located at the neuromuscular junction and is the consequence of an antibody-mediated autoimmune reaction against the postsynaptic acetylcholine receptors (AChR) in skeletal muscle (110). By contrast, the autoimmune response in Eaton-Lambert syndrome, a myasthenic syndrome seen almost exclusively in adults harboring a malignancy (most commonly a small cell carcinoma of the bronchus), is directed against the voltage-sensitive calcium channels on the presynaptic membrane.
Antibodies to the AChR protein have been demonstrated in the serum of a large proportion of patients with generalized juvenile myasthenia gravis (111), and the electrophysiologic features of the disease can be reproduced in animals by repeated injections of IgG derived from myasthenic subjects or by antibodies against the AChR (112). Several lines of evidence indicate that antibody production against AChR is T-cell dependent. Serum antibodies against AChR are usually absent in congenital myasthenia gravis (but not in the maternally transmitted neonatal form) and in many cases of myasthenia, that is limited to extraocular muscle involvement. A significant proportion of the small number of juvenile myasthenics who are AChR antibody negative have antibodies against a receptor tyrosine kinase (MuSK). MuSK has been localized to the neuromuscular junction and appears to be vital to its in utero development (112,113). Children and adults with anti-MuSK antibodies exhibit weakness of extraocular, pharyngeal and respiratory muscles and later should have shoulder girdle weakness. Plasma exchange and prednisone often are effective treatments, but cholinesterase inhibitors and thymectomy appear ineffective (113a). Some patients with congenital myasthenia gravis have been demonstrated to have anti-MuSK antibodies (113b). Still other congenital myasthenic infants with episodic apnea show mutations in the cholin acetyltransferase (CHAT) gene (113c).
By means of snake venom (bungarotoxin, which binds specifically to the AChR), AChRs have been localized to the postsynaptic folds. Myasthenic subjects show a 70% to 90% reduction in the number of functional AChR (114). The gross destruction of the AChR area, seen in subjects with long-standing myasthenia gravis, is probably the consequence of a cellular immune reaction. Three possible processes have been postulated to be operative: a complement-mediated lysis of the postsynaptic membrane, an IgG-induced accelerated rate of degradation by endocytosis of cross-linked acetylcholine receptors, or an antibody-mediated blockage of the active site of the receptors (112,114).
The factors that induce and sustain an autoimmune reaction against the AChR are still unknown, although there are some hints about the nature of the process. Administration of penicillamine to patients with rheumatoid arthritis can induce myasthenia that is reversible within a few months after withdrawal of the medication. Whereas this observation suggests an alteration of the AChR because of exogenous factors, Lindstrom and coworkers believe that patients with myasthenia gravis respond to some endogenous source of native AChR, rather than to a bacterial or viral coat protein that would not have the identical autoantibody specifications (110).
The initial anti-AChR sensitization probably occurs in the thymus. Extracts of thymic tissue have been found to contain acetylcholine receptors that are localized to myoid cells present in that tissue (115), and thymocytes of myasthenic patients produce AChR antibodies (116). In most instances, these antibodies are specific for the embryonic AChR (117). Whereas an environmental factor might initiate an immune response against the AChR in the thymus of genetically predisposed subjects, examination by immunofluorescence of thymus derived from patients with myasthenia gravis does not show these cells to be the foci of immunologic stimulation (118). Alternatively, a possibly viral-induced derangement of function or communication between the various types of cells that regulate the intensity and duration of an immune reaction (immune regulation) might be responsible for the breakdown in tolerance to AChR protein.
What appears most likely at present is that autoimmune myasthenia gravis is a heterogeneous group of diseases distinguishable by their clinical features, notably the age at onset of the illness and by their human leukocyte antigen types, with each entity having a different pathogenetic mechanism. Thus, in some 10% to 15% of adult myasthenic patients, antibodies against AChR cannot be detected, and evidence suggests that in these subjects, antibodies are directed to other parts of the end-plate, such as MuSK, rather than to the AChR (119). The immunopathology has been reviewed by Hughes and colleagues (120).
Whereas juvenile myasthenia gravis, like adult myasthenia gravis, appears to result from T-cell initiated antibodies directed against end-plate AChR protein, neonatal myasthenia is associated with the transfer of maternal AChR antibodies across the placenta. However, the story is not as simple as this. Maternal AChR antibodies are found in all infants of myasthenic mothers, whether infants are symptomatic or not, and myasthenic symptoms in an infant are not proportional to the amount of antibodies transferred to it from the mother (121,122). Rather, neonatal myasthenia appears to correlate with persistence of antibodies, possibly as a consequence of antibody synthesis by the myasthenic infant.
TABLE 16.5 Myasthenic Conditions of Infancy and Childhood | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Clinical Manifestations
Before noon, the stores of the spirit which influenced the muscle being almost spent, they are scarcely able to move hand and foot…. [T]his person for some time speaks freely and readily enough, but after long, hasty or laborious speaking, presently she becomes mute as a fish and cannot bring forth a word.
—T. Willis (109)
Neonatal myasthenia is a transient disease seen in approximately one in five infants born to mothers who generally are suffering from active acquired myasthenia gravis (125). Symptoms usually appear during the first 24 hours or, at the latest, by the third day of life. All affected infants have a paresis of the lower bulbar muscles, causing a weak cry and difficulty in sucking or swallowing. Generalized hypotonia is found in approximately one-half of the infants. Antibody titers against the AChR are elevated in the mother and in most but not all infants. Symptoms respond promptly to anticholinesterase medication, and even if untreated, the illness usually lasts less than 5 weeks.
Congenital myasthenia gravis designates children with myasthenia born to mothers without the disease (126,127). Generally, antibodies against the AChR protein are undetectable. Several conditions have been recognized. These are outlined in Table 16.5 (126,127,128,129,130,131,132,133,134,135,136,137). Harper and Engel and coworkers have divided these into presynaptic, synaptic, and postsynaptic defects; primary AChR kinetic abnormalities; and the slow-channel congenital myasthenic syndrome (126,127).
Presynaptic defects include at least two autosomal recessive conditions caused by defects in acetylcholine resynthesis and mobilization (127,138,139). Postsynaptic defects include several forms associated with defects in the gene that codes for the collagenlike tail of acetylcholine esterase (131,140), and more than 60 mutations in one of the five genes coding for the adult or fetal subunits of the acetylcholine receptor (127). These entities are depicted in Table 16.5.
The most common congenital myasthenic syndrome is caused by a kinetic abnormality of AChR, the slow-channel syndrome. This is an autosomal dominant condition and is the consequence of an abnormally prolonged open-time of the acetylcholine-ion channels (127,135,141). Less common are the various mutations that cause the fast-channel syndrome. In these mutations, the postsynaptic
response to acetylcholine is markedly diminished, although the number of acetylcholine receptors per end-plate is normal (136,142). The numerous genetic defects that have been demonstrated in congenital myasthenic syndromes were recently summarized in tabular form, with references provided (143).
response to acetylcholine is markedly diminished, although the number of acetylcholine receptors per end-plate is normal (136,142). The numerous genetic defects that have been demonstrated in congenital myasthenic syndromes were recently summarized in tabular form, with references provided (143).
TABLE 16.6 Symptoms and Signs in 35 Patients with Juvenile Myasthenia Gravis | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
The clinical picture of congenital myasthenia also is heterogeneous. In approximately one-half of children, symptoms commence before 2 years of age, and more than one sibling can be affected (124,139). In many instances, fetal movements are reduced, and neonates can have feeding difficulties, ptosis, limitation of eye movements, and a weak cry. The initial symptoms in congenital myasthenia gravis are not as severe as in the neonatal variety, and the diagnosis is, therefore, more difficult. A few patients with congenital myasthenia have spontaneous remissions, but the course of the disease is usually protracted, with mild symptoms that often are refractory to both medical and surgical therapy (thymectomy).
Myasthenia can begin during childhood (juvenile myasthenia). In this form, girls are affected two to six times as frequently as boys. The onset of juvenile myasthenia can be insidious, although at times, it is rapid and often a sequel to an acute febrile illness. Generally, muscles innervated by the cranial nerves are affected first, with bilateral ptosis the most common presenting sign (Table 16.6 and Fig. 16.3). It was seen in 81% of children in the series of Afifi and Bell and was unilateral in 33% (111). Generalized weakness and dysphagia are less common presenting symptoms. The clinical course is highly variable. Approximately one-half of the patients experience one or more remissions, usually during the early years of the illness. In others, symptoms progress to a certain point and then become stationary. In 26% of cases in the series of Afifi and Bell, myasthenia was restricted to the extraocular musculature (111).
The characteristic feature of all forms of juvenile myasthenia gravis is the variability in muscular strength, with increasing weakness of the affected muscles with repeated contractions (Fig. 16.4). The child is usually at his or her best in the morning and becomes progressively weaker during the day, although partial or complete recovery may follow a nap. In approximately 10% to 20% of untreated cases, weakness becomes irreversible, and muscle wasting, particularly of the shoulder girdle and the extraocular muscles, becomes apparent. Despite hypotonia, tendon jerks are normal or even exaggerated, but they may disappear after repeated elicitation.
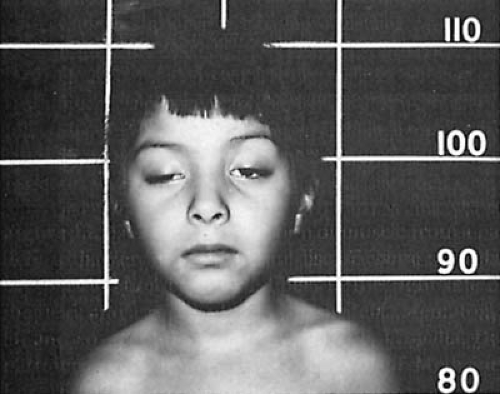 FIGURE 16.3. Girl with myasthenia gravis. She has bilateral ptosis, more marked on the right, and the typical myasthenic facies. (Courtesy of Dr. Christian Herrmann, Jr., Department of Neurology, University of California, Los Angeles, UCLA School of Medicine.) |
Other autoimmune diseases are sometimes associated with juvenile myasthenia gravis. The most common of these are rheumatoid arthritis, juvenile diabetes mellitus, and thyroid disease, usually thyrotoxicosis (144). For reasons as yet unexplained, seizures are seen in as many as 12.5% of patients (145,146). Malignancies were seen in 5%
of the Mayo Clinic pediatric series (147). Postinfectious myasthenia gravis in children is transitory and usually follows a varicella-zoster infection in 2 to 5 weeks as an immune response (147a).
of the Mayo Clinic pediatric series (147). Postinfectious myasthenia gravis in children is transitory and usually follows a varicella-zoster infection in 2 to 5 weeks as an immune response (147a).
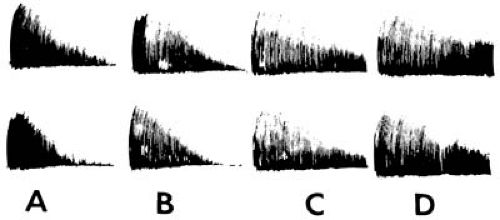 FIGURE 16.4. Muscle ergograph in a patient with myasthenia gravis. A: Resting state. B: After intravenous atropine (0.4 mg). C: After intravenous Tensilon (2.0 mg). D: After intravenous edrophonium chloride (8.0 mg). Note the progressive decrease in amplitude in A and B, partly or completely corrected by edrophonium chloride. (Courtesy of Dr. C. Herrmann, Jr., Department of Neurology, University of California, Los Angeles, UCLA School of Medicine.) |
Myasthenia also can be seen in association with collagen vascular disease, notably systemic lupus erythematosus. Dobkin and Verity and Rodolico and others have described a familial limb girdle myasthenic syndrome transmitted through an autosomal recessive gene and characterized by cardiomyopathy, excessive muscle fatigability with a predilection for the proximal musculature, and improvement with anticholinesterase drugs (148,149). Muscle biopsy revealed hypoplasia of type 1 muscle fibers and tubular aggregates of types 1 and 2 fibers.
Diagnosis
The diagnosis of myasthenia gravis is based on a history of abnormal fatigability of voluntary muscles and evidence of a prompt improvement of muscle weakness with anticholinesterase drugs. It is supported by a history of remissions and exacerbations and by the absence of any concurrent illness that might contribute to the muscular weakness.
For diagnostic purposes, the most widely used drug is edrophonium chloride (Tensilon). Approximately 5 mg is given intravenously to a 3- to 5-year-old child. Within 1 minute after the injection, a transient improvement in muscle strength is observed, usually lasting 4 to 5 minutes. Positive responses are diagnostically reliable. In some infants, a positive response can be obscured by the response to the injection, in which case a longer-acting anticholinesterase drug such as neostigmine should be used. After intramuscular injection of 0.5 to 1.5 mg of neostigmine, muscle strength begins to improve in 10 to 15 minutes and reaches its peak by 30 minutes. Both tests are nearly 100% reliable in generalized myasthenia as well as in ocular myasthenia, which progresses to generalized myasthenia. Neostigmine should never be administered intravenously because it may induce fatal cardiac arrythmias. Edrophonium should be avoided in infancy for the same reason. In the series of Afifi and Bell, edrophonium gave positive test results in all cases of ocular myasthenia; neostigmine was positive only in 67% of cases (111). A number of conditions can give a false-positive result. These include drug-induced myasthenia gravis, botulism, brainstem gliomas, and Guillain-Barré syndrome (154). A confirmatory test, therefore, is required.
The presence of AChR antibodies in serum is the simplest diagnostic test in juvenile myasthenia, although false-negative results are not unusual, particularly in children whose symptoms are limited to the extraocular muscles. In the series of Afifi and Bell, AChR antibodies were found in 63% of cases (111). A similar incidence of positive serology has been reported in other series. When AChR antibodies are absent, it is necessary to consider one of the congenital myasthenic syndromes. This is not an academic exercise because in juvenile myasthenia gravis, therapy is not only supportive but also is directed against the immune-mediated response. Andrews and coworkers suggest that the rapid response to plasmapheresis frequently seen in children with juvenile myasthenia gravis can be used to distinguish this condition from congenital myasthenia gravis (155). It should be noted that antibody levels do not parallel disease severity.
Rapid stimulation of the distal (ulnar) or proximal (spinal, axillary, or facial) motor nerves produces a characteristic decrement in the amplitude of successive action potentials recorded from surface muscle electrodes and in the amplitudes of the mechanical excursions (see Fig. 16.4). This test is positive in some 80% of pediatric cases, but is usually negative in ocular myasthenia (156). Patients suspected of having myasthenia gravis on the basis of their clinical picture but who have a normal EMG study should undergo single-fiber EMG (157,158). The results of this study generally complement the results of the assay for AChR antibodies. Exceptions are seen in the various syndromes of congenital myasthenia gravis, in which single-fiber EMG is frequently positive; another exception is in children with the mild, predominantly ocular form of the disease (158).
In addition to penicillamine, a number of other drugs can induce or unmask myasthenia. These include β-adrenergic blockers, carnitine, aminoglycoside antibiotics, lithium carbonate, magnesium salts, trimethadione, and phenytoin (159).
Treatment
Treatment of the patient with juvenile myasthenia is generally lifelong, extending into all areas of activity. The three approaches to therapy are symptomatic, immunologic, and surgical. Treatment of the congenital myasthenias is solely symptomatic (160). As a rule, the first step is remission induction. This is usually accomplished through the use of high-dose corticosteroids, frequently in conjunction with intravenous immunoglobulin or plasmapheresis. Maintenance of the remission is then usually accomplished by slow tapering of the corticosteroids along with the use of “steroid-sparing” agents, which include thymectomy and immunosuppressants.
When juvenile myasthenia is mild or is restricted to the ocular musculature, treatment is generally symptomatic (i.e., using anticholinesterase drugs). At the University of California, Los Angeles, UCLA School of Medicine, pyridostigmine bromide (Mestinon) is the preferred drug. It
is administered at a starting dosage of 15 mg two to three times a day (in tablet or syrup form) for a child 3 to 8 years old, or 0.5 to 1.0 mg/kg.
is administered at a starting dosage of 15 mg two to three times a day (in tablet or syrup form) for a child 3 to 8 years old, or 0.5 to 1.0 mg/kg.
In the remaining patients, treatment should be as outlined above. Mann and coworkers and Vajsar suggest a course of oral prednisone, starting out at high dosages (1 mg/kg per day) and tapering when improvement is sustained or when side effects require reduction of the dosages (161,162). Sarnat and associates have advocated a starting dosage of 2 mg/kg given every other day (163). Cholinesterase inhibitors are used as needed while the patient is receiving corticosteroids. Exacerbation of myasthenia gravis can be seen during the early days of corticosteroid therapy, usually toward the end of the first week, and can last approximately 1 week. Improvement is noted within approximately 2 weeks and becomes maximal in approximately 3 months. Plasmapheresis lowers the level of antibodies against the acetylcholine receptor protein. Dau recommends this procedure to stabilize patients whose course worsens markedly during initiation of prednisone therapy and as primary therapy in combination with immunosuppressive drugs such as azathioprine (164). Plasmapheresis also has been used to treat patients who remain unresponsive to other forms of therapy, to improve respiratory function before thymectomy, and to hasten remission after thymectomy. Improvement, often dramatic, is seen within 1 to 3 days after the start of the course. Immunosuppression of refractory patients with azathioprine (2 to 3 mg/kg), cyclosporine, and cyclophosphamide (50 mg/kg per day) has been disappointing because of side effects such as bone marrow suppression and hepatotoxicity (162,165) and is not used at our institutions. Intravenous immune globulin has been used in conjunction with other modes of therapy, particularly corticosteroids and plasmapheresis, in rapidly deteriorating patients with bulbar symptoms (166). There is little difference between the efficacy of three plasma exchanges and intravenous immune globulin (0.4 mg/kg per day for 3 to 5 consecutive days), although as a rule, intravenous gammaglobulin is better tolerated (167). Intravenous immune globulin is ineffective in neonatal myasthenia gravis (168).
After establishment of maximal improvement, children should be considered for thymectomy (166,169,170), although the effectiveness of the procedure in the pediatric population has not been validated by controlled trials. In the German series of Lindner and coworkers, 82% of children were thymectomized (146). In general, the procedure is now indicated if symptoms recur after withdrawal of corticosteroids after 1 year of therapy or if corticosteroids fail to benefit the patient significantly. The best results from thymectomy may be predicted in children who have high titers of circulating antiacetylcholine receptor antibodies and who are symptomatic for less than 2 years. However, a long history of juvenile myasthenia gravis is not a contraindication to thymectomy; for both girls and boys, good operative results are possible. In the UCLA experience, the procedure is of considerable benefit when performed as early as the third year of life. The maximum benefit of thymectomy may be delayed for weeks or even several months, so absence of an immediate and dramatic postoperative improvement should not be discouraging.
Proceeding according to this treatment schedule, Mann and associates found that up to 90% of patients have significant improvement or complete remission of their myasthenic symptoms and that in some, corticosteroids and anticholinesterase drugs can even be tapered and discontinued (161). In the series of Lindner and colleagues, 60% of patients who underwent thymectomy went into remission, as compared with 29% of those who were not thymectomized (146). In the series of Rodriguez and coworkers, the remission rate was higher in children who underwent thymectomy within 12 months of the onset of symptoms. It also was higher in patients who had experienced bulbar symptoms before surgery, in those having involvement of extremities but neither ocular nor generalized weakness, and in those whose myasthenic symptoms presented between ages 12 and 16 years (147).
Patients with congenital forms of myasthenia may respond to anticholinesterases. When such treatment is unsatisfactory, Palace and colleagues have suggested 3,4-diaminopyridine. This substance blocks potassium conductance and increases the release of acetylcholine from the nerve terminal (171).
Toxic Disorders
The toxins of Clostridium tetani and C. botulinum and the venom of cobras and of arachnoideae such as the black widow spider affect the neuromuscular junction. These toxins are more extensively considered under their respective headings in Chapter 10.
DISEASES OF MUSCLE
Muscular Dystrophies
The muscular dystrophies are a group of diseases that are distinguished from other neuromuscular disorders by four obligatory criteria: (a) primary myopathies, not neurogenic; (b) genetically determined; (c) progressive diseases, some slowly progressive and compatible with normal longevity and others more rapidly progressive and leading to early death; and (d) myofiber degeneration at some stage in the disease (5).
An excellent review on the history of the terminology in muscular dystrophy was provided by Dubowitz (172). The name muscular dystrophy, meaning a growth disorder of muscle, was entrenched by Gowers in his “Clinical lecture on pseudohypertrophic muscular paralysis,” a series of essays in The Lancet in 1879 (173).
From a clinical point of view, there are at least eight major forms of muscular dystrophy: Duchenne
(Aran-Duchenne) muscular dystrophy, Becker muscular dystrophy (now known to be merely a mild form of Duchenne dystrophy with the same genetic defect), Emery-Dreifuss (humeroperoneal) muscular dystrophy, facioscapulohumeral (Landouzy-Déjèrine) muscular dystrophy, the limb girdle dystrophies, oculopharyngeal muscular dystrophy, distal muscular dystrophy, and the congenital muscular dystrophies. The characteristic genetic and clinical features of each entity are presented in Table 16.7 (174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200).
(Aran-Duchenne) muscular dystrophy, Becker muscular dystrophy (now known to be merely a mild form of Duchenne dystrophy with the same genetic defect), Emery-Dreifuss (humeroperoneal) muscular dystrophy, facioscapulohumeral (Landouzy-Déjèrine) muscular dystrophy, the limb girdle dystrophies, oculopharyngeal muscular dystrophy, distal muscular dystrophy, and the congenital muscular dystrophies. The characteristic genetic and clinical features of each entity are presented in Table 16.7 (174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200).
Pathologic Anatomy
Despite differences in the clinical picture, the same pathologic findings are shared by all muscular dystrophies. The earliest stages of the disease show a dilated sarcoplasmic reticulum, with an irregular orientation of the triads (201). As the illness progresses, there are repeated episodes of necrosis and regeneration of muscle cells. This process is probably initiated by a breakdown of the muscle cell membrane, and electron microscopy may reveal the absence of the plasma membrane around all or part of the circumference of the muscle fibers (202). Small losses of membrane can be repaired, but generally, regeneration is inadequate. As a consequence of the membrane defect, an influx of calcium ions can occur, activating the endogenous proteases and inducing lysis of the Z-disc of the myofibril, which is probably the initial step in muscle breakdown. Subsequently, the number of muscle cells is reduced, and variability in fiber size is increased. Sarcolemmal nuclei are swollen and increased in number, a regenerative reaction to the injury of muscle fibers. Over the years, this reaction becomes more extreme, and muscle fibers appear enlarged, forked, hyalinized, or atrophied. With further progression of the disease, large accumulations of collagen and fat cells are seen between muscle fibers, and these cells are partly responsible for the muscular hypertrophy (Fig. 16.5) (45).
Neither heart nor smooth muscle is spared, and there is myocardial degeneration with fatty infiltration and fibrosis of the myocardial fibers.
When muscular dystrophy is associated with mental retardation, brain sections can be normal or can disclose various developmental anomalies of the cortex, such as pachygyria, heterotopia, and disorders of cortical architecture (203).
Pathogenesis
The elucidation of the gene defect in Duchenne and Becker muscular dystrophy represents one of the most important triumphs of molecular biology in the area of neurology, and was the first human disease whose nature was clarified by positional cloning (173). The gene for Duchenne and Becker muscular dystrophy is localized on Xq21.2. It is 10 times larger than any other gene characterized to date and consists of approximately 2 million base pairs; its product has been named dystrophin, a protein with a molecular weight of 427,000. The most common gene mutation in both Duchenne and Becker muscular dystrophies is a deletion. The phenotype of Duchenne and Becker muscular dystrophies does not always correlate with the size of the deletion in the dystrophin gene; some cases of undetectable dystrophin are paradoxically associated with only mild clinical manifestations (204). Rather, the effect of the deletion on dystrophin synthesis is important. If the deletion breaks up the codon triplet and thereby shifts the reading frame, a premature stop codon is quickly generated, and dystrophin synthesis comes to a halt, leaving a small, truncated protein molecule without its carboxy terminal, which is rapidly degraded. If the deletion removes a triplet codon in its entirety, the reading frame is maintained, resulting in the synthesis of a dystrophin molecule with a shortened rod domain but intact carboxy and amino domains. This is what occurs in Becker dystrophy (205).
Dystrophin is a low-abundance protein and constitutes but 0.002% of total muscle protein. It is a cytoskeletal protein with a globular amino domain, a central rodlike domain, and a globular carboxy domain. It is localized to the inner surface of the sarcolemma, aggregating as a homotetramer that is associated with actin at its amino terminus and with a glycoprotein complex at its carboxy terminus. The dystrophin-glycoprotein complex, by which the membrane cytoskeleton is linked to extracellular glycoproteins, consists of several dystrophin-associated glycoproteins and dystrophin-associated proteins (Fig. 16.6). The membrane complex is markedly reduced in Duchenne muscular dystrophy. Its synthesis is normal, but in the absence of dystrophin, the glycoproteins become unstable and are degraded (206). These associated glycoproteins include not only the dystroglycans and sarcoglycans, but also the syntrophin-dystrobrevin complex that is highly localized at the neuromuscular synaptic membrane and is later to appear in normal fetal development (206a).
Dystrophin is present not only in muscle, but in a variety of other cell types, including brain and retina (207,208,209). Although in situ hybridization demonstrates some expression of the gene in affected muscle (210), the complete dystrophin molecule is absent or is present in less than 3% of normal levels in more than 90% of patients with classic Duchenne muscular dystrophy. It is present in reduced amounts or in normal amounts but with an abnormal molecular configuration in boys with Becker muscular dystrophy (211).
The level of dystrophin expression in brain is lower than in muscle, and the structure of brain messenger RNA for dystrophin differs from that of muscle (212,213). This difference is because cortical dystrophin is transcribed from a promoter that is at least 90 kb upstream from the muscle promoter. Other, markedly truncated dystrophins can be found in normal brain and peripheral nerve. In particular, a 140-kD protein (Dp140) is found throughout the CNS (214,215).
TABLE 16.7 Clinical and Genetic Features of the Muscular Dystrophies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
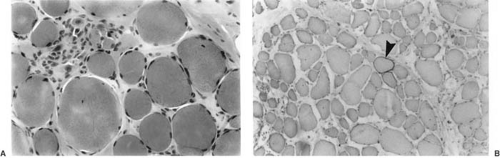 FIGURE 16.5. Muscle biopsy of a 5-year-old boy with Duchenne muscular dystrophy. A: Myofibers are extremely variable in size with both hypertrophic and atrophic forms. They have an abnormally rounded contour rather than the normal polygonal contour in the transverse section because of the proliferation of endomysial collagen that forms rigid sleeves around each fiber. A few fibers have internal nuclei. A zone of degenerating myofibers infiltrated by phagocytic cells is seen in the upper left of the figure. B: Immunocytochemistry shows absence of dystrophin at the sarcolemma of all myofibers, large and small, except for one (arrowhead) called a revertant fiber that expresses this protein as in normal fibers (A, hematoxylin and eosin, × 400; B, dystrophin, rod domain, × 250). |
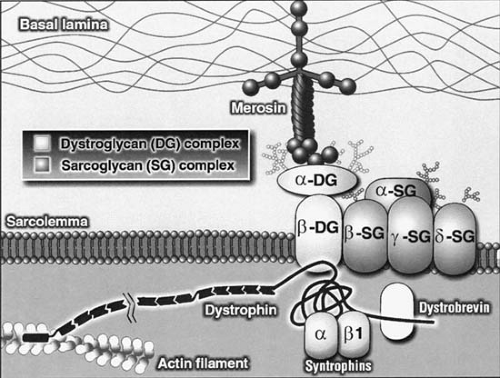 FIGURE 16.6. The dystrophin-glycoprotein complex. Dystrophin contacts F-actin in the cytoplasm of the cell, and the dystrophin-glycoprotein complex forms a bridge across the membrane to the merosin subunit of laminin in the extracellular matrix. (Courtesy of R. G. Worton, The Ottawa Hospital, Research Institute, Ottawa.) |
Dystrophin is associated with other proteins at the sarcolemmal region, which also may be defective; these include α- and β-dystroglycans (216,217,218,219) and utrophin. Utrophin is an autosomal homologue of dystrophin, being encoded by a gene that has been mapped to the long arm of chromosome 6 (6q24). Like dystrophin, it is a large protein that is found at the neuromuscular synapse and the myotendinous junction and also is expressed in several other tissues, notably the lungs and intestines (220).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


