Biogenic amines
Dopamine
Regulation of motor activity and in integrative aspects of behavior, cognitive functions, and attention-related processes
Norepinephrine
Regulation of vigilance, reactivity, and executive function
Serotonin (5-hydroxytryptamine)
Modulation of tonic of nervous system activity, feeding behavior, and circadian rhythms. Precursor of the melatonin
Histamine
Control of biological rhythms, behavioral state, energy metabolism, thermoregulation, fluid balance, and reproduction
Amino acids
Glutamate
Excitatory in the central nervous system. Regulation of sensorimotor functions, cognitive functions, emotions, and memory
γ-amino butyric acid (GABA)
Inhibitory in the central nervous system
Glycine
Inhibitory in the brainstem and spinal cord, co-agonist of glutamate on NMDA (excitatory) receptors
Acetylcholine
Regulation of all motor activity, alertness, cognitive functions, emotions, and memory
2 Neurotransmitters and Their Metabolites
2.1 Tetrahydrobiopterin Metabolism
Tetrahydrobiopterin (BH4) is a cofactor of phenylalanine, tyrosine, tryptophan hydroxylases, and all nitric oxide synthases (Blau et al. 2001). BH4 plays an essential role in the synthesis of L-dopa and 5-hydroxytryptophan from tyrosine and tryptophan, respectively (the initial and rate limiting step in the biosynthesis of biogenic amines) (Blau et al. 2001; Hyland 1999).
BH4 provides electrons during reactions catalyzed by phenylalanine, tyrosine and tryptophan hydroxylases, and is oxidized to its hydroxyl compound – pterin-4α-carbinol-amine which is further converted to quininoid dihydropterin (qBH2) by pterin-4 α-carbinolaminedehydratase (PCD; EC 4.2.1.96) and next regenerated to BH4 by dihydrobiopterin reductase (DHPR; EC 1.6.99.7) (Blau et al. 2001).
The synthesis of BH4 involves three main reactions. The first step of the BH4 synthesis is conversion of guanosine triphosphate (GTP) to dihydroneopterin triphosphate (NH2P3) by rate-limiting enzyme GTP cyclohydrolase I (GTPCH I; E 3.5.4.16). GTPCH activity is regulated by the regulatory protein GFPR and phenyloalanine concentrations. A defect in GTPCH I exists in both autosomal recessive (arGTPCH) and dominant (adGTPCH) forms. The second enzyme is 6-pyruvoyl-tetrahydropterin synthase (PTPS; EC 4.6.1.10) that converts dihydroneopterin triphosphate (NH2P3) to 6-pyruvoyl-tetrahydropterin (6-PTP). As the last step, sepiapterin reductase (SR; EC 1.1.1.153) reduces 6-PTP to BH4. DHPR is an enzyme of regeneration of BH4 from qBH2 (Blau et al. 2001; Hyland 1999).
Defects in the biosynthesis of BH4 may occur with or without hyperphenylalaninemia (HPA). HPA is usually detected through the neonatal screening program and about 2 % of HPA detected by the newborn screening are due to disorders in BH4 metabolism (Blau et al. 2001). DHPR, PTPS, and arGTPCH I deficiencies are common in patients with HPA and can be differentiated by pterin profile in the urine and CSF, DHPR activity in the blood, and a BH4 loading test. PTPS deficiency increases the level of neopterin, along with normal or decreased of biopterin in the CSF and urine. DHPR usually leads to increased biopterin in the urine and CSF. ArGTPCH I causes a decrease in both neopterin and biopterin in the urine and CSF (Pearl et al. 2006; Blau et al. 2001). The autosomal dominant form of GTPCH deficiency, commonly known as the Segawa syndrome or dopa-responsive dystonia, and SR deficiency are not connected with HPA and can be detected only in CSF (Friedman et al. 2012; Kusmierska et al. 2009; Pearl et al. 2006).
The most frequent symptom of ad GTPCH deficiency is dopa-responsive dystonia with diurnal fluctuation. The major symptoms of the SR deficiency are motor delay, axial hypotonia, dystonia, and oculogyric crises with diurnal fluctuation of symptoms (Friedman et al. 2012)
There is a ‘salvage pathway’ for sepiapterin metabolism by its reduction by carbonyl reductase (CR) or SR to 7,8-dihydrobiopterin (BH2) that can be further reduced to BH4 by active dihydrofoliate reductase (DHFR). DHFR is active only in the liver, and therefore patients with SR deficiency do not have HPA and have a normal pterin profile in the urine. In the central nervous system (CNS), DHFR is not active, so that the ‘salvage pathway’ is not active in the brain and a deficit of SR leads to decreased concentrations of BH4 and biogenic amine metabolites in CSF (Blau et al. 2001). SR deficiency cannot be detected by conventional methods applied for the measurement of biogenic monoamine metabolites in CSF, as it must be distinguished from all pterins in CSF, especially BH2 and sepiapterin. The detection method is based on the finding of a marked decrease in SR activity in fibroblasts, along with SR gene (SPR gene) molecular analysis (Friedman et al. 2012; Kusmierska et al. 2009).
2.2 Biogenic Amines Metabolism
Tyrosine hydroxylase (TH; EC 1.14.16.2), aromatic L-amino acid decarboxylase (AADC; EC 4.1.1.28), dopamine β-hydroxylase (DβH; EC 1.14.17.1), and monoamine oxidase (MAO; EC 1.4.3.4) deficiencies are the known deficiencies of biogenic amine metabolism (Hyland 1999).
The TH is the rate-limiting enzyme in the biosynthesis of catecholamines (dopamine, norepinephrine, and epinephrine) (Fig. 1). TH deficiency leads to decreased concentration of HVA in CSF. 5-HIAA should be normal, but sometimes, a slight decrease of 5-HIAA is observed. A low concentration of HVA without any other biochemical disturbances also can suggests an adGTPCH I deficiency. A phenylalanine loading test is helpful to differentiate these two defects. TH deficiency can be confirmed only by molecular analysis (Blau et al. 2001; Hyland 1999). The most characteristic clinical symptoms of TH deficiency are psychomotor retardation, extrapyramidal symptoms (dystonic and choreoathetotic movements, and parkinsonian symptoms), and an oculogyric crisis (Hoffmann et al. 2003).
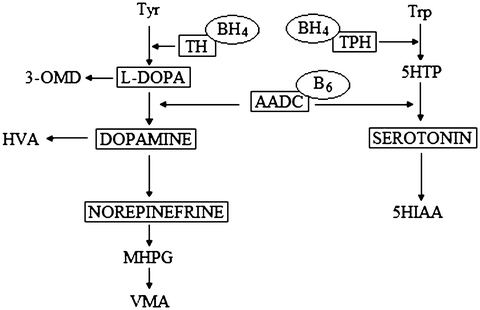
Fig. 1
Schematic presentation of biogenic amine metabolism. Tyr tyrosine, Trp tryptophan, TH tyrosine hydroxylase, TPH, tryptophan hydroxylase, BH4 tetrahydrobiopterin, 5HTP 5-hydroxytryptophan. 5HT 5- hydroxytryptamine, AADC aromatic L-amino acid decarboxylase, 5HIAA 5-hydroxyindoleacetic acid, MHPG, 3-methoxy-4-hydroxyphenylglycol, HVA homovanillic acid, VMA, vanillylmandelic acid
The aromatic L-amino acid decarboxylase (AADC) enzyme is the last enzyme in the chain of dopamine and serotonin synthesis. AADC deficiency is connected with decreased concentrations of HVA and 5-HIAA and increased concentrations of 3-O-methyl-dopa (3-OMD) and 5-hydroxytryptophan (5-HT) in CSF. Additionally, in AADC deficiency vanillactic acid (VLA) and vanillpyruvic acid (VPA) occur in the urine, which may be detected by gas chromatography–mass spectrometry (GC-MS). AADC deficiency can also occur with decreased concentration of 5-methyltetrahydrofoliate (5-MTHF) (Clayton 2006). The characteristic clinical symptoms of AADC deficiency are similar to those of TH deficiency above outlined. In addition, there may be truncal hypotonia, limb hypertonia, body temperature instability, and hypoglycemia (Ide et al. 2009; Swoboda et al. 2003). A mutation in SLC18A2 gene, which encodes VMAT2, results in a phenotype which overlaps with monoamine disorders (Rilstone et al. 2012).
2.3 Foliate Metabolism
Folic acid is not a neurotransmitter, but it is closely linked to neurotransmitters’ metabolism (Hyland et al. 2010). Foliate is a main cofactor in DNA biosynthesis, necessary for the methylation process. It is a critical determinant of the embryonic central nervous system development and participates in the synthesis of purines, pyrimidines, and in the metabolism of serine, histidine, methionine, and glycine. Folic acid also affects the activity of some enzymes, such as DHPR, AADC, and 3-phosphoglycerate dehydrogenase. 5-methyl-tetrahydrofolate (5-MTHF) is decreased in CSF of patients with a low folic acid concentration. Folic acid has three separate transporter systems (Hyland et al. 2010). Deficiency of the primary transporter may lead to cerebral foliate deficiency (CFD) and decreased 5-MTHF concentration in CSF. A defective secondary system may be confirmed by increases in HVA and 3-OMD in CSF.
2.4 γ-Aminobutyric Acid (GABA) Metabolism
GABA is a major inhibitory neurotransmitter of the brain and is formed from glutamic acid by the B6-dependent enzyme – glutamate decarboxylase (GAD; EC4.1.1.15). GABA is transported into synaptic vesicles by the vesicular GABA transporter (VGAT). GABA is converted to succinic semialdehyde (SSA) in the reaction catalyzed by GABA transaminase (GABA T; EC 2.6.1.19) (Jakobs et al. 1981). Then, succinic semialdehyde is oxidized by succinic semialdehyde dehydrogenase (SSADH; EC 1.2.1.24) to succinate that is successively used in Krebs cycle. Two defects of the GABA metabolism have been reported: GABA-T deficiency and SSADH deficiency (Jaeken et al. 1984; Jakobs et al. 1981).
The best known neurotransmitter disorder is SSADH deficiency. The main symptoms of SSADH deficiency are a developmental delay and intellectual disability, behavioral problems, motor dysfunction (ataxia), and epilepsy (Kim et al. 2011). GABA-T deficiency is a rare disorder of GABA catabolism, with a severe psychomotor retardation and recurrent episodic lethargy accompanied by intractable seizures (Tsuji et al. 2010).
2.5 Pyridoxine-Dependent Enzymes
Vitamin B6 is present in the organism as six vitamers: pyridoxine (pyridoxol), pyridoxamine, pyridoxal, and their 5′-phosphorylated esters. The biologically active form of pyridoxine (cofactor activity) is pyridoxine-5-phosphate (PLP) which is a cofactor for approximately 100 enzymes. Pyridoxine dependency is caused by the binding of PLP to abnormal metabolites formed due to the deficiency of alpha-aminoadipic semialdehyde dehydrogenase. AADC also depends on the PLP; thus a PLP disorder affects the biogenic amine metabolite profile in CSF in a way similar to the primary low AADC activity. PLP deficiency occurs with increased glycine concentration in CNS, causing neurological disturbances. Lack of PLP is the most commonly caused by decreased pyridoxine-5-phosphate oxidase (PNPO) activity. PNPO deficiency leads to severe neurological sequelae. A typical clinical presentation is characterized by perinatal onset, epileptic encephalopathy, refractoriness to antiepileptic drugs, and responsiveness to PLP treatment (Rahman et al. 2012; Clayton 2006; Pearl et al. 2006; Jaeken et al. 1984; Jakobs et al. 1981).
Related posts:
 Health Status Influence Acceptance of Illness in Patients with Chronic Respiratory Diseases?
Functioning of the Prelingually Deaf Adults
Health Status Influence Acceptance of Illness in Patients with Chronic Respiratory Diseases?
Functioning of the Prelingually Deaf Adults
 Protects Glial Cells Against 6-Hydroxydopamine Toxicity
Introduction
Protects Glial Cells Against 6-Hydroxydopamine Toxicity
Introduction
 E
E
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


