tips and tricks
- Distal myopathies are frequently confused with inherited sensorimotor neuropathies.
- Consider a distal myopathy when:
there is insidious onset of distal weakness without sensory symptoms
the extensor digitorum brevis muscles are intact in bulk while anterior leg compartment muscles are atrophic.
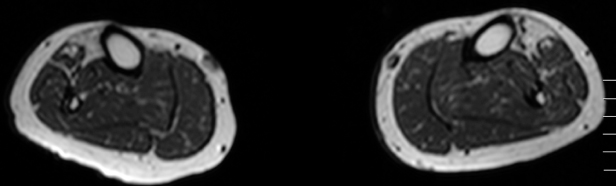
When a decision is taken that one is dealing with a distal myopathy, the pattern of weakness and mode of inheritance may further help narrow the diagnosis to a fraction of the list shown in Table 12.1. However, ultimately, a specific diagnosis will rely on histopathological findings on muscle biopsy and confirmation of the diagnosis by genetic testing. As different muscles can be very differently affected in distal myopathies, with pathology ranging from normal to end-stage, the site of biopsy has to be carefully evaluated. Muscle magnetic resonance imaging (MRI) is the preferred method to target which muscle will likely give the optimal yield on pathology. Moreover, MRI provides excellent additional diagnostic clues due to the distinct patterns of muscle involvement in the different distal myopathies.
After MRI and muscle biopsy (Figure 12.1), molecular genetic testing is needed to establish a final diagnosis. Distal dysferlinopathy (Miyoshi myopathy) can be diagnosed by protein immunohistochemistry and western blotting of the muscle sample, and the group of myofibrillar myopathy causing distal myopathies can be assessed by staining with antibodies against the accumulating mutant proteins such as desmin, myotilin, and αB-crystallin. See Table 12.1 for the list of currently identified distal myopathies.
Figure 12.1. Muscle magnetic resonance imaging shows highly selective fatty degeneration of tibialis anterior and minor early changes in long toe extensors in distal titinopathy (Udd myopathy, TMD).
Clinical Features
Welander distal myopathy usually causes reduced finger extension after the age of 50, followed by lower leg weakness and finger flexor weakness. Onset of symptoms in the lower legs occurs in a minority of patients. Walking is preserved and lifespan is intact.
Tibial muscular dystrophy (Udd myopathy or TMD) starts with ankle dorsiflexion weakness after the age of 30–50, with slow progression to moderate foot drop 10–15 years after onset. Walking is usually preserved until age 85 and there is no reduction of lifespan.
Distal myotilinopathy also starts even later, usually after the age of 50, showing reduced plantar flexion and in some patients dysphonia. The progression is, however, more rapid and may lead to loss of ambulation within 10 years.
Zaspopathy (Markesbery–Griggs type) is very similar to distal myotilinopathy, although first symptoms are both dorsal and plantar flexion weakness, the progression is slower, including atrophies of intrinsic hand muscles, and cardiomyopathy may develop at late stages.
Matrin-3 mutated VCPDM (vocal cord and pharyngeal distal myopathy) is very late onset with ankle dorsiflexion weakness, foot drop, and clinical symptoms of dysphonia and dysphagia as additional features. The progression is slow.
VCP (valosin-containing protein)-mutated myopathies usually cause proximal or scapuloperoneal phenotypes. Nevertheless, in some families scapular or proximal involvement is not present and, in such cases with distal muscle weakness only, the clinical phenotype may be indistinguishable from Udd or Welander myopathy.
Desminopathy has an earlier onset than the previous disorders, starting usually before the age of 30 years. The first symptoms and signs are either cardiac weakness or distal weakness in the ankles. Progression to proximal muscles and severe disability usually occur within 10 years. Cardiomyopathy may be present many years before the skeletal myopathy, and involvement of respiratory muscles is common.
Patients with the distal myopathy of the type described in the Australian family from Victoria had first signs of weakness in the hand grip as young adults. There was slow progression to thenar atrophy and posterior calf atrophies, with subsequent plantar flexion weakness without total loss of ambulance even in the late 70s.
Laing distal myopathy is very early onset; sometimes reduced ankle dorsiflexion is present from the first years of walking. Slow progression to finger extension weakness and neck flexor weakness is the rule, whereas late generalized muscle weakness and severe disability are unusual. Cardiomyopathy may occur but is rare. So-called new mutations are frequent, which means that many patients present as sporadic cases without any family history.
Miyoshi myopathy (MM) was long thought to have a pathognomonic phenotype with early adult onset of calf muscle atrophy and weakness combined with very high CK levels, usually 50–100 times the upper normal limit. The disease progression is moderate with involvement of proximal muscles and disabilities 10–15 years after onset.
A subset of patients may present with a similar phenotype to MM, albeit with no loss of dysferlin protein in the muscle biopsy (dysferlin is mutated in MM). Recently another gene defect, anoctamin-5, was identified to cause a similar clinical phenotype, with the exception that MM is usually very symmetric whereas anoctaminopathy frequently causes asymmetric muscle involvement.
Nonaka distal myopathy also presents in early adulthood, with clear ankle dorsiflexion weakness as the first sign. The progression to involvement of proximal muscles is moderate to severe, causing loss of ambulation 12–15 years after the onset of symptoms.
There are, however, other myopathies that may present with distal weakness that can be mistaken for distal myopathies. Myotonic dystrophy type 1 (DM1) typically causes distal weakness in the hands and ankles, with no major proximal limb muscle weakness in the early stages of the disease. However, when distal weakness is present, the DM1 patient usually shows facial weakness and myotonia, leading to the correct diagnosis. Late-onset cases of sporadic inclusion body myositis (s-IBM) may mimic a distal myopathy because one hallmark of the disease is finger flexor weakness. In s-IBM, distinct quadriceps weakness and atrophy should be present. Quadriceps atrophy in s-IBM is in direct contrast to the quadriceps sparing in hereditary inclusion body myopathy/Nonaka distal myopathy (HIBM), which is an early adult-onset, autosomal recessive form of distal myopathy. Facioscapulohumeral muscular dystrophy (FSHD) usually presents with characteristic weakness and atrophy of facial and proximal upper limb muscles, with scapular winging, but it may rarely present with ankle dorsiflexion weakness with no obvious scapular or facial weakness.
 tips and tricks
tips and tricks
- Muscle MRI is extremely valuable for assessment of the pattern of muscle involvement as well as for targeting the optimal muscle for biopsy.
- Of the distal myopathies, only desminopathy causes major involvement of heart and respiratory muscles.
- Many underlying gene defects occur as new mutations and therefore lack any family history.




