4
EEG Interpretation in Childhood Epilepsies
In this chapter we address the electroencephalography (EEG) of pediatric epilepsies. The epilepsies of premature babies or neonates are beyond the scope of this chapter.
PAROXYSMAL DISCHARGES AND SEIZURE DIAGNOSIS
In evaluating the EEG of a patient with possible seizures, we may see interictal epileptiform discharges (IEDs) and/or nonspecific paroxysmal discharges, with or without focal or diffuse slowing. IEDs, represented by spike or spike-wave discharges, are the most sensitive and specific markers for the diagnosis of seizures. In a routine EEG (a recording of approximately 30 min) the chance of recording a clinical seizure (ictal) event is rather rare, unless the patient is having frequent seizures or is in status epilepticus. Thus, we often rely primarily on IEDs for the diagnosis of epilepsy.
The likelihood of detecting IEDs varies, depending on seizure type, age, and seizure frequency. An EEG that includes sleep or is recorded after sleep deprivation increases the yield of IEDs. Generally, greater seizure frequency is associated with higher yield of IEDs (1). IEDs are also recorded more often in children than in adults. Detection of IEDs differs, depending on the origin of the epileptiform activity: If a relatively small area of cortex is involved as the epileptogenic zone, IEDs may not be detected by scalp electrodes. Also, epileptiform activity arising from deep brain structures such as the medial temporal lobe, subfrontal lobe, or interhemispheric medial cortex may not be readily recorded by scalp electrodes.
The specificity of IEDs is determined by the incidence of IEDs in the normal population (false-positive), compared with that in patients with epilepsy. IEDs are found in 1.9% to 3.5% of healthy children (2,3) and 0.5% of healthy adults (4). Specificity also varies depending on the type of IEDs: Only about 40% of the patients with benign rolandic spikes of childhood or benign epilepsy of childhood with centrotemporal spikes (BECTS) and 50% of patients with childhood epilepsy with occipital paroxysms (benign occipital spikes of childhood) have a history of seizures (5). Also IEDs elicited by photic stimulation and generalized spike-wave discharges are less correlated with seizure history compared to focal spikes. Multifocal IEDs and focal IEDs, especially at the midline, frontal, and anterior temporal regions are highly (75%–95%) correlated with clinical seizures (5,6). Overall, the incidence of detecting IEDs during the first EEG in adult epilepsy patients is about 30% to 50% (7,8). In children less than 10 years old, the incidence is about 80%. Overall, repeating the EEG once increases the yield an additional 20% to 30% (7,8).
THE EPILEPSIES
Certain forms of epileptic seizure disorders have special clinical and EEG characteristics irrespective of their etiologies. The International Classification of Epilepsies and Epileptic Syndromes were proposed by the Committee on Classification and Terminology of the International League Against Epilepsy (9). This classification is based on two principles, distinguishing first between localized (focal) and generalized epilepsies and second between idiopathic and symptomatic etiologies.
Normal or near-normal background activity is characteristic of idiopathic epilepsy, and slowing of background activity or multifocal epileptiform activity are suggestive of symptomatic epilepsy. Focal background abnormality and/or focal polymorphic delta activity are likely correlated with symptomatic epilepsy.
Localized (Focal) Epilepsies and Syndromes
In the localized epilepsies and syndromes, examples of the idiopathic type include benign childhood epilepsy with centrotemporal spikes (rolandic spikes or BECTS and childhood epilepsy with occipital paroxysms (benign childhood occipital lobe epilepsy). Symptomatic localized epilepsies and syndromes include frontal, parietal, and temporal or occipital lobe seizures that are secondary to focal pathology.
The localization of an epileptogenic focus often determines the character of the seizures. To some extent, it is possible to speculate on the clinical manifestation of seizures based on the localization and waveform of epileptiform activity. Conversely, it may be possible to postulate the localization and waveforms of epileptiform activity based on clinical seizure types.
Benign Epilepsy of Childhood With Centrotemporal Spikes
Spikes maximally recorded from central or midtemporal electrodes have been described as BECTS or benign rolandic epilepsy. The waveform is more commonly “sharp” rather than a true spike, having a triphasic configuration with a prominent negative peak, preceded and followed by small positive peaks. The negative field centered at the central or midtemporal electrode is commonly associated with positive fields in the frontal region (Figure 4.1A–B). These discharges are often unilateral but one-third of the patients have bilateral independent foci. Sleep (non–rapid eye movement [non-REM]) greatly increases the spikes and about one-third of the patients have spike discharges only in sleep (10). Also, unilateral spikes in the awake state may progress to bilaterally independent spikes in sleep. Rolandic spikes tend to decrease progressively with age and eventually disappear by the midteen years (11). The onset of seizures usually occurs between 4 years and adolescence. Consistent with the dramatic increase of spike discharges in sleep, about 80% of seizures occur exclusively in sleep (12).
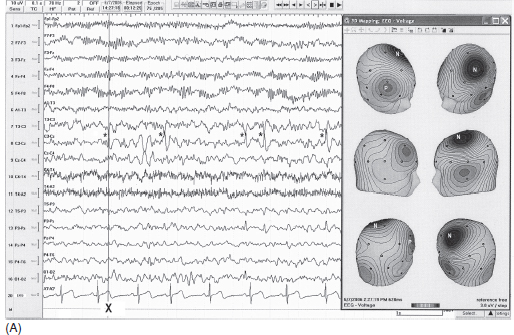
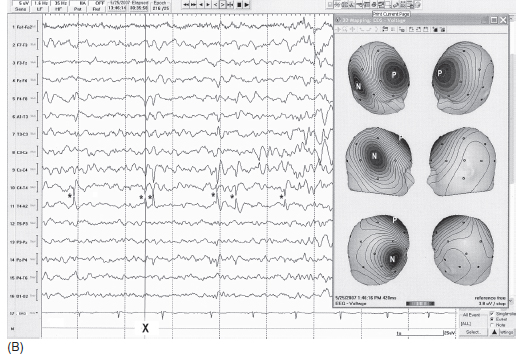
FIGURE 4.1 (A) Benign childhood epilepsy with centrotemporal spikes (BECTS) in a 12-year-old girl with a history of nocturnal generalized tonic-clonic convulsions with postictal aphasia and right arm weakness (Todd’s paralysis). Electroencephalogram (EEG) showed spikes maximum at C3 (marked by *). Note the maximum negative (N) field at C3 and positive (P) field at contralateral frontal regions shown by three-dimensional (3D) topographic mapping (maps were made at the negative peak marked by X). (B) Benign childhood epilepsy with centrotemporal spikes (BECTS) in a 10-year-old boy with a history of nocturnal generalized tonic-clonic convulsions. Electroencephalogram (EEG) showed spikes maximum at T4 (marked by *). Note the maximum negative (N) field at T4 and positive field (P) at contralateral frontal region shown by three-dimensional (3D) topographic mapping (maps were made at the negative peak marked by X).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Clinically, seizures initially consist of unilateral paresthesias of tongue, lips, cheek, and gum and/or unilateral tonic-clonic activity of facial and pharyngeal/laryngeal muscles contralateral to the side of the spike focus. The ictal discharges often start with an initial electrodecremental pattern followed by rhythmic spike bursts with subsequent spike-wave discharges. The seizure ends without postictal slowing, unless the seizure evolves to a generalized tonic-clonic convulsion, which is not uncommon.
Not all spike discharges from central regions are benign. Some features help to distinguish benign rolandic epilepsy from symptomatic (nonbenign) epilepsy. As shown in Figures 4.1A–B, benign rolandic spikes are tangentially oriented with a negative field just behind the rolandic fissure, either at or close to the central or midtemporal electrode, and a positive field over the frontal region. In symptomatic epilepsy, spikes have often radially oriented distribution; thus the negative field spreads diffusely over a wide scalp region (12). Focal slowing (corresponding to the side of spikes) is absent in benign rolandic epilepsy but it is often present in symptomatic epilepsy (Figure 4.2). When a rolandic spike is maximum at the midtemporal electrodes, differentiation of temporal lobe epilepsy with spikes at T3/T4 electrode from benign rolandic epilepsy can be more difficult. In this case, additional electrodes between C3/C4 and T3/T4 (C5/C6 according to the expanded 10-20 system) help to distinguish the two: Rolandic spikes are maximum at C5/C6, whereas symptomatic temporal lobe seizures likely have a maximum at T3 or T4 (13).
Childhood Epilepsy With Occipital Spikes
There are two types of seizures categorized in this entity: late onset, originally described by Gastaut (14) (Gastaut type) and early onset, more recently described by Panayiotopoulos (15), and now referred to as Panayiotopoulos syndrome. Affected children are neurologically normal in both types. Family history of epilepsy is positive in more than one-third of the patients for the late-onset type but negative for the early-onset type. In the late-onset variant, age of seizure onset ranges from 15 months to 17 years with a peak age of 7 to 9 years (16). In the early-onset variant, onset is between 1 and 14 years with a peak age of 3 to 6 years (17).
In the late-onset type, seizures almost always begin with visual symptoms (blindness, scintillating scotoma, visual hallucinations, or illusions). The seizure is usually brief, lasting only a few to several seconds. Half of the patients complain of severe migraine-like headache associated with nausea and vomiting (14). Thus, differentiation from migraine headache may be sometimes difficult. The prognosis is good overall but less favorable than that of BECTS.
In the early-onset type, seizures lack characteristic visual symptoms but consist of a variety of autonomic symptoms including “feeling sick,” paleness, nausea, vomiting, cyanosis, myosis, or mydriasis, and cardiopulmonary irregularities. The seizure lasts much longer (5–10 min) than the late-onset type and often ends with a hemiconvulsion, Jacksonian march, or generalized motor activity. About one-third of the seizures occur in sleep. Nearly all patients become seizure-free by age 12 years (17).
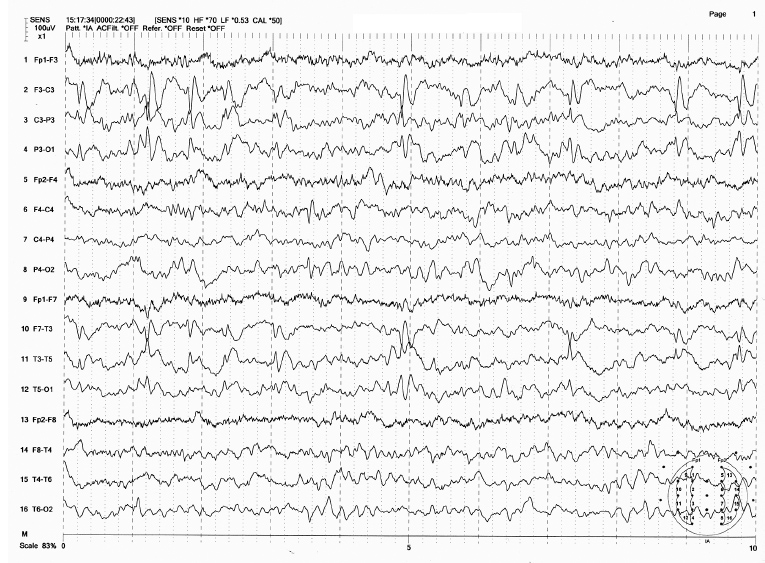
FIGURE 4.2 Spike discharges at left central and midtemporal region in a 6-year-old boy with a history of focal seizure involving the right arm. Note the spike discharges maximum at left central and midtemporal region and also the increased delta and decreased background activity over the left hemisphere. This is unlikely to be benign childhood epilepsy with centrotemporal spikes (BECTS).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
EEG abnormalities are indistinguishable between the Gastaut or the Panayiotopoulos type of seizures. Spikes consist of high-amplitude surface negativity (200–300 µV), often followed by a small positive and negative slow wave (Figure 4.3). These occur singly or more commonly in serial, semirhythmic bursts with a unilateral or bilaterally independent appearance. Eye opening tends to abolish the spike wave and eye closing may precipitate the burst. However, neither repetitive photic stimulation nor hyperventilation precipitates epileptiform discharges (16). As with BECTS, sleep (non-REM) increases the discharges, but less prominently as compared to BECTS.
About one-fourth to one-third of the patients have other epileptogenic abnormalities including generalized spike-waves or BECTS (16,18) (Figure 4.4). The ictal EEG consists of rhythmic spike discharges starting from one occipital region evolving into rhythmic theta-delta that spreads to the contralateral occipital region (Figure 4.5).
Benign childhood occipital lobe epilepsy must be differentiated from other occipital spikes. Many patients with congenital or acquired amblyopia are found to have occipital spikes, but the morphology of the spike is much faster (“needlelike spike”) than that of benign childhood occipital epilepsy (19). Occipital spikes may also occur in idiopathic generalized seizure patients with photoparoxysmal response (20) (Figure 4.6A–B). Patients may have localization-related seizures with vivid visual hallucinations (21). Although many “pure” simple partial seizures fail to show ictal discharges, partial seizures of occipital origin tend to show well-localized ictal EEG activity and the patient often is able to describe his/her visual hallucinatory event in detail.
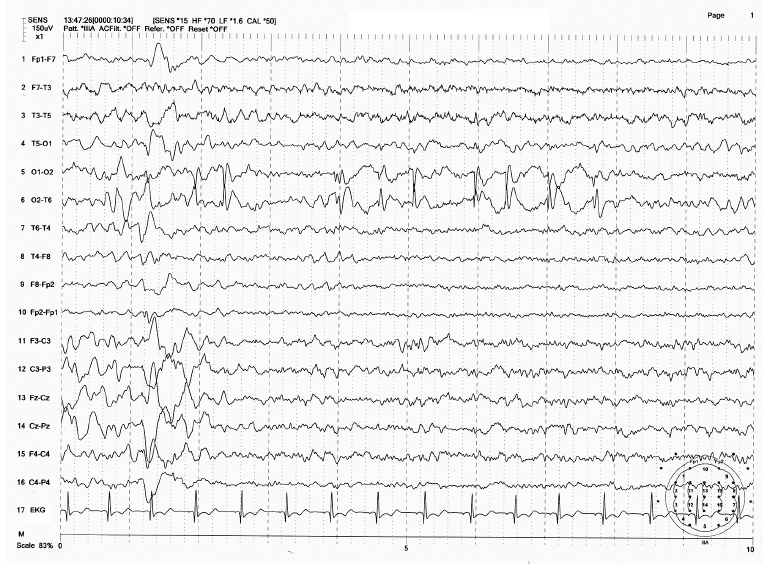
FIGURE 4.3 Childhood epilepsy with occipital paroxysms (benign occipital lobe epilepsy) in a 4-year-old boy whose seizures manifest as seeing “flashing light.” Note the high-amplitude occipital spikes at O2 electrode.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Generalized Epilepsies
Idiopathic generalized epilepsies and syndromes include absence seizures (childhood and juvenile) and juvenile myoclonic epilepsy (JME). West syndrome and Lennox-Gastaut syndrome (LGS) are examples of generalized epilepsy of a symptomatic type.
Primary Generalized Epilepsies
Generalized epileptiform discharges appear in both hemispheres simultaneously with similar configuration, symmetric amplitude, and synchronous timing between homologous electrodes. Timing between anterior and posterior discharges may differ slightly within the same hemisphere. When discharges consistently have a higher amplitude in one hemisphere or generalized discharges are consistently preceded by focal discharges, this suggests a focal onset to a secondarily generalized seizure type, but clear-cut differentiation between a primary and secondary generalized seizure pattern is not always clear-cut. Distinction between ictal and interictal patterns in absence seizures is also not as clear as that of a focal seizure. Often, the EEG associated with a clinical seizure may be simply a longer and more rhythmic repetition than interictal IEDs.
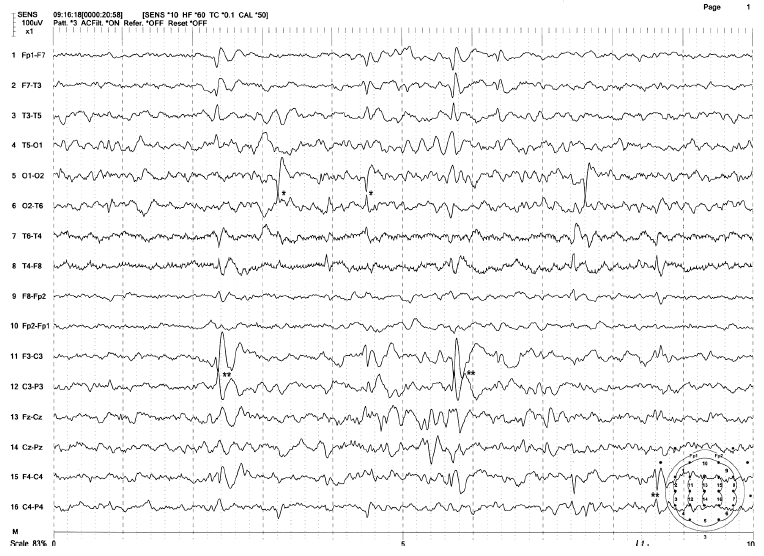
FIGURE 4.4 Two coexisting types of benign paroxysm—benign rolandic spike of childhood (BECTS) and childhood epilepsy with occipital paroxysm (benign occipital childhood epilepsy) in a 10-year-old girl with a history of generalized tonic-clonic seizures. Note the two independent BECTS at C3/T3 and C4/T4.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
ABSENCE EPILEPSY The discovery of 3 Hz spike-wave bursts in association with petit mal or absence seizures by Gibbs, Davis, and Lennox (22) in 1935 was the first major epoch in the history of EEG and the electrographic diagnosis of epilepsy. This is characterized by rhythmic cycles of spike-wave complexes at a frequency of about 3 Hz, usually lasting a few to several seconds. The spike-wave complexes are generally maximum in the frontal region and may start with a frequency 3 to 3.5 Hz and end with a frequency 2 to 3 Hz (Figure 4.7). The initial complex may have a polyspike-wave pattern. Although the bursts seem to be synchronous between the two hemispheres, detailed analysis with a faster sweep recording shows that the spikes in one hemisphere may randomly precede those in the other hemisphere by a few milliseconds (23). The discharges tend to be inhibited by eye opening or increased vigilance. Hyperventilation often precipitates the bursts associated with clinical absence seizures in 50% to 80% of the patients (24). Intermittent photic stimulation induces spike-wave bursts in about one-fifth of the patients with absence epilepsy (25). Non-REM sleep also increases the number of spike-wave complexes, which tend to become more irregular with polyspike-wave patterns (26). REM sleep decreases the number of bursts to a frequency slightly less than the waking state (26). Valproic acid and ethosuximide, commonly used medications for absence seizures, tend to decrease the number of spike-wave bursts (27) and also attenuate activation by photic stimulation (28).
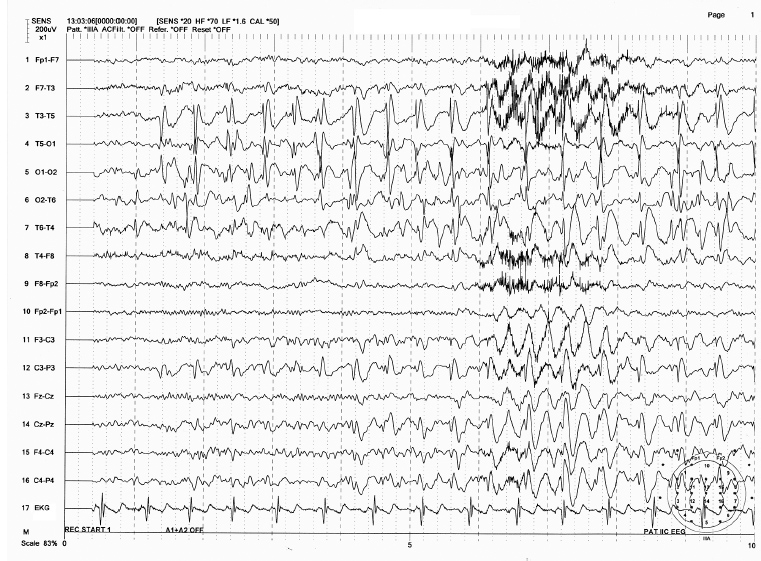
FIGURE 4.5 Ictal discharges in childhood epilepsy with occipital paroxysm (benign occipital childhood epilepsy) in an 11-year-old girl with ictal manifestation of blindness, nausea, and migraine-like headache. Note the rhythmic spike-wave discharges initially originating from O1 electrode with subsequent spread to other electrodes. Also there were independent spikes at T6 electrode, which became synchronized with the occipital spikes as the seizure progressed.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
In patients with absence epilepsy, background activity is usually normal, but slowing can occur in a minority of patients. Generally, about 20% to 40% of patients with absence seizures show 3 Hz rhythmic delta bursts in the occipital regions (occipital intermittent rhythmic delta activity [OIRDA]) (Figure 4.8), and the incidence of OIRDA is much higher in children between 6 and 10 years (29). Visible clinical seizures can usually be observed when spike-wave bursts last more than 4 to 5 seconds. Symptoms are characterized by staring, impaired responsiveness, and behavior arrest. Impaired responsiveness can occur with spike-wave bursts as short as 3 seconds. Responsiveness returns abruptly to normal at the end of the spike wave. Detailed psychophysiological testing of such patients found decreased reaction time even during a brief spike-wave burst without an associated overt clinical seizure (30). Automatisms with lip smacking, chewing, fumbling, or mild myoclonus (eyelid twitches) resembling a temporal lobe seizure may be seen (31). The frequency of eyelid twitches is usually 3 Hz coinciding with 3 Hz spike waves. Some patients may have prominent eyelid myoclonus with faster frequency of 4 to 10 Hz. This is referred to as Jeavons syndrome (eyelid myoclonia with absences) (32). Some patients may have decreased postural tone. It is the technologist’s role to examine the presence or absence of impaired responsiveness and to note the patient’s behavior changes during spike-wave bursts. Absence seizures are classified into two types based on the age of onset.
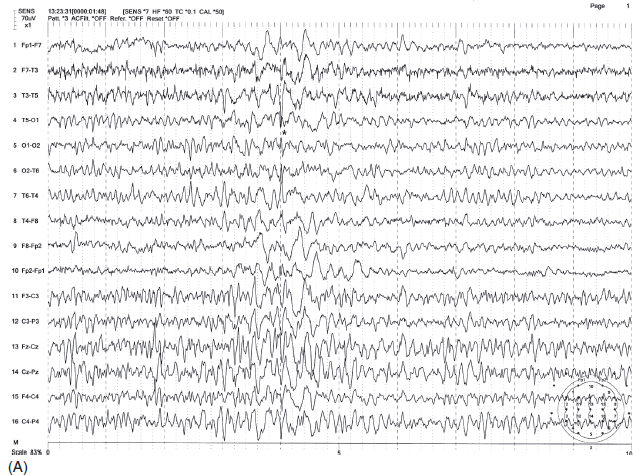
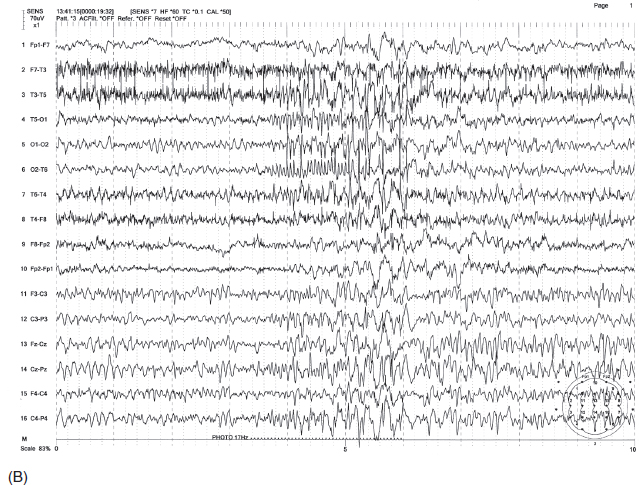
FIGURE 4.6 (A) Occipital spikes in a 34-year-old man with a history of generalized tonic-clonic convulsions since childhood. Note the spike discharges are maximum at O1 electrode (indicated by *). (B) This patient also had a photoparoxysmal response at 17 Hz with the initial occipital polyspikes time-locked to the flashes, followed by generalized irregular spike-wave bursts.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
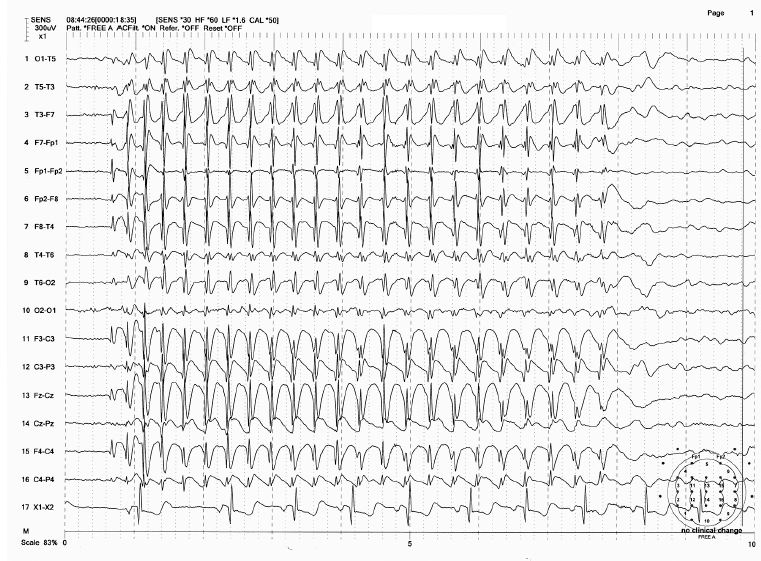
FIGURE 4.7 Spike-wave bursts of 3 Hz associated with absence seizure in a 7-year-old boy (childhood absence). Note that spike-wave bursts started with a frequency slightly faster than 3 Hz and ended with a frequency slightly slower than 3 Hz.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Childhood Absence Epilepsy Onset of epilepsies is from 3 to 12 years. EEG and clinical presentation can assist in predicting the prognosis. Patients with an EEG showing 3 Hz OIRDA have a smaller risk of developing tonic-clonic seizures in the future (33). Absence seizures without myoclonus have a higher chance of remission than those with myoclonus (34).
Juvenile Absence Epilepsy The onset of juvenile absence epilepsy (JAE) is around 10 to 12 years or even later. The spike-wave bursts tend to be of a slightly faster frequency than 3 Hz and may have polyspike components (Figure 4.9). This group of patients is more likely to develop generalized tonic-clonic seizures or myoclonic seizures (35).
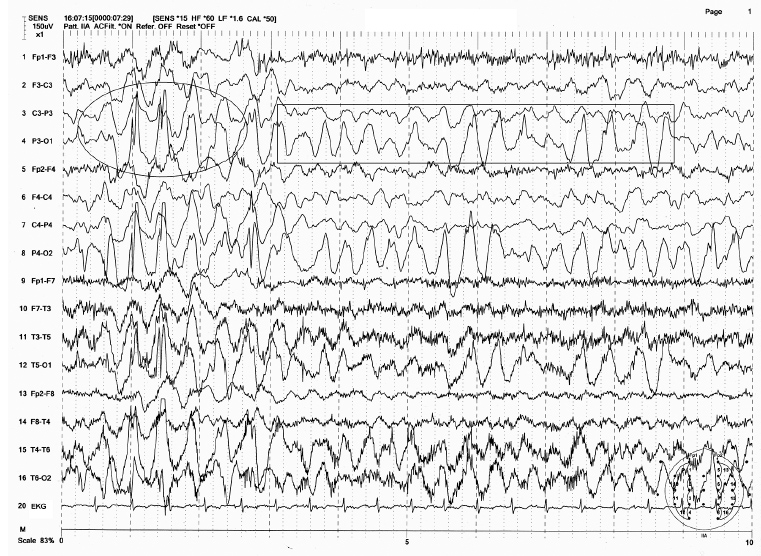
FIGURE 4.8 An example of occipital intermittent rhythmic delta activity (OIRDA) in a 9-year-old boy with a history of absence seizures. Note the occipital dominant 3 Hz spike-wave burst (shown by oval circle) mixed with 3 Hz OIRDA (shown by rectangular box).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
GENERALIZED TONIC-CLONIC EPILEPSIES Multiple spike-wave (polyspike-wave) bursts lasting less than 1 to several seconds are usually an interictal expression of a generalized seizure (Figure 4.10A–B). The interictal EEG of idiopathic (primary) generalized tonic-clonic epilepsies consists of a variety of waveforms, which are more irregular and of a faster frequency than the 3 Hz rhythmic spike-wave discharges seen in absence seizures. In addition, spike-wave bursts often include multiple spikes (polyspike). Spike-wave bursts are usually, but not always, symmetric and synchronous. Asymmetric bursts, however, do not necessarily exclude the possibility of primary generalized epilepsy. In fact, it is often difficult to differentiate primary from secondary generalized epilepsy, especially in the case of frontal lobe epilepsy. In some patients, focal spike discharges, such as “rolandic spikes,” may coexist with generalized spike-wave bursts. In such cases, it is not possible to determine if the patient has a seizure of focal onset with secondary generalization or has both partial and primary generalized epilepsy. There are two features, which may help point toward a diagnosis of primary generalized epilepsy. One is a photoparoxysmal response (Figure 4.11A–B), and the other is generalized spike-wave bursts resembling K-complexes (Figure 4.12A–B). Like K-complexes, spike-wave bursts act as an arousal pattern. In some, generalized spike-wave bursts may be precipitated by arousal stimuli, similar to the K-complex. In some patients, especially in children who tend to have “spiky” K-complexes, the differentiation between the two could be difficult. These features were studied in detail by Niedermeyer, who introduced the concept of “dyshormia” in which primary generalized spike-wave bursts and K-complexes share the same generating mechanism producing generalized burst activity (36).
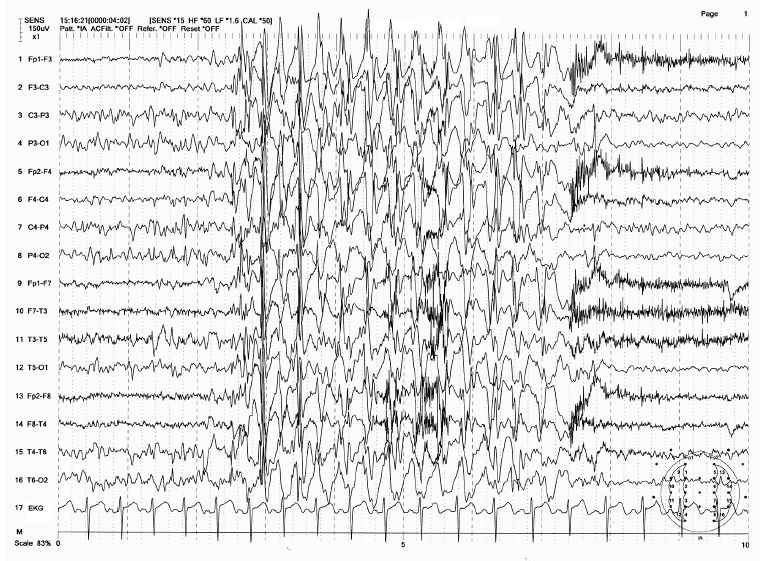
FIGURE 4.9 Spike-wave bursts of 3 Hz with initial polyspike component in an 18-year-old girl (juvenile absence). The patient had absence seizures in the past and recently started having generalized tonic-clonic convulsions.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Onset of the ictal event in a generalized seizure consists of low-voltage, rhythmic beta-range fast activity, with progressively increasing amplitude and decreasing frequency (Figure 4.13A–B). This is followed by generalized spike-wave bursts, which become progressively slower in frequency and less rhythmic toward the end of the seizure. The EEG becomes suppressed during the immediate postictal period and is then followed by the appearance of postictal delta activity. Clinically, the initial fast activity corresponds with the tonic phase and the subsequent spike-wave bursts coincide with the clonic phase of the seizure. During the ictal events, EEG activities are largely obscured by muscle and movement artifacts, making it difficult to differentiate a genuine seizure from a pseudoseizure. The presence of postictal flattening or slowing provides evidence of a genuine seizure. Conversely, immediate normalization of the EEG favors a pseudoseizure, especially when the patient is unconscious or confused.
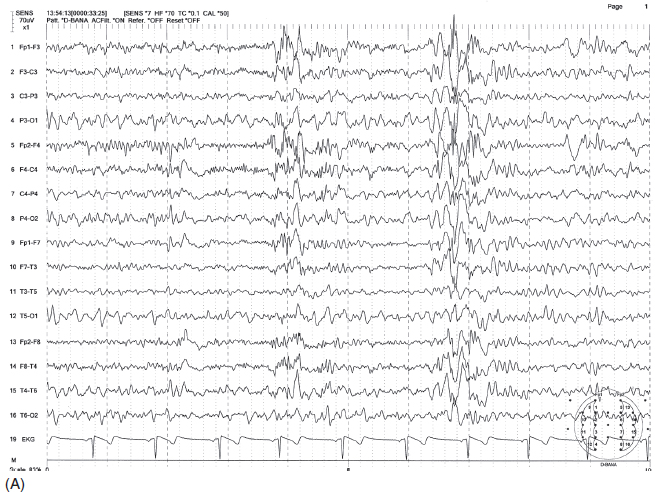
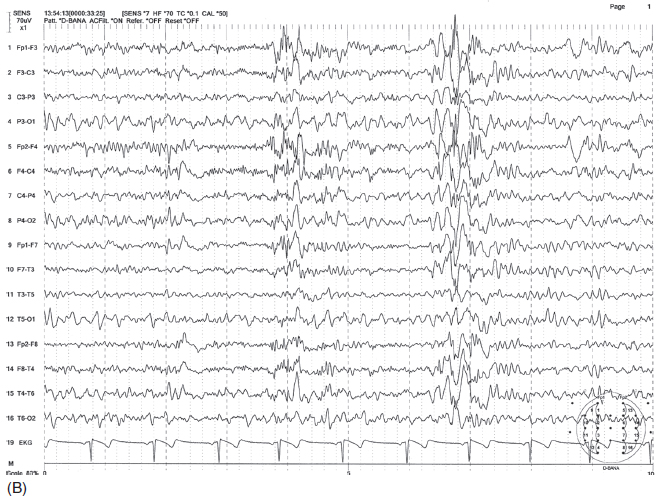
FIGURE 4.10 Somewhat irregular, bilaterally synchronous generalized spike-wave bursts of 4 to 6 Hz, maximum at midline (A) in a 30-year-old woman with a history of generalized tonic-clonic convulsions as well as myoclonic seizures since childhood. The patient also had more irregular polyspike-wave bursts during stage two sleep (B).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
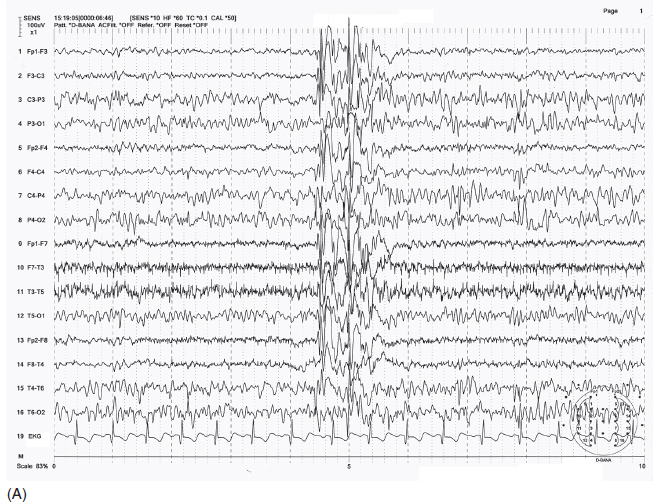
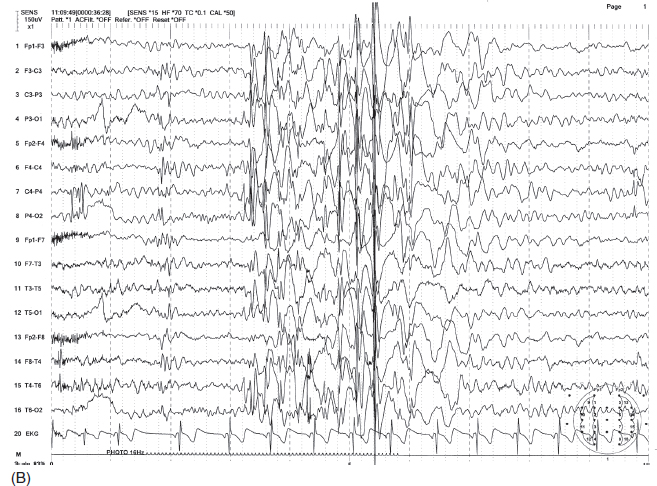
FIGURE 4.11 Generalized irregular spike-wave bursts (A) in a 10-year-old boy with a history of absence seizures and a recent grand mal seizure. Photic stimulation produced photoparoxysmal response at 16 Hz frequency flashes with generalized irregular spike-wave bursts (B).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
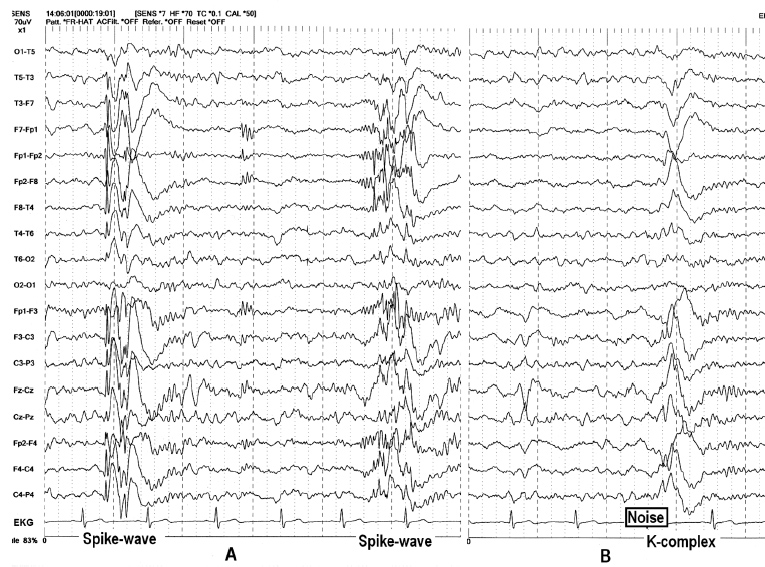
FIGURE 4.12 (A) Bilaterally diffuse synchronous and symmetric spike-wave bursts with bifrontal dominance in a 31-year-old man with a history of grand mal seizures since childhood. Note that the spike-wave bursts were followed by spindles. (B) With the exception of the spikes, the epileptiform bursts had similar waveform and distribution with K-complex induced by noise.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
JUVENILE MYOCLONIC EPILEPSY This was originally described by Janz as impulsive petit mal (37) and may be referred to as juvenile myoclonic epilepsy of Janz. JME is the most common type of seizure among idiopathic generalized epilepsies. Close to half of the patients have a family history of epilepsy (37). As the name implies, seizures usually begin in adolescence. The majority of patients (>90%) also have generalized tonic-clonic convulsions. Both myoclonic and generalized tonic-clonic convulsions tend to occur within 1 to 2 hours after awakening. A history of absence seizures coexists with or precedes myoclonic seizures in about one-third of the patients (38).
Interictal EEG patterns consist of generalized polyspike and polyspike-wave discharges with frontocentral predominance (Figure 4.14A). These patterns are not distinguishable from other idiopathic generalized epilepsies but may include more polyspike components. Like other idiopathic seizures, background activity is usually normal. Spike-wave or polyspike-wave bursts are usually faster than the typical 3 Hz spike-waves seen in absence seizures, but in some patients, 2.5 to 3 Hz spike-wave bursts may occur that are indistinguishable from typical absence seizures.
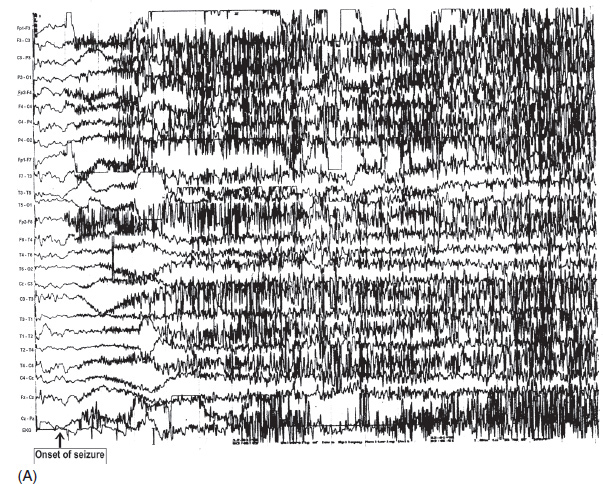
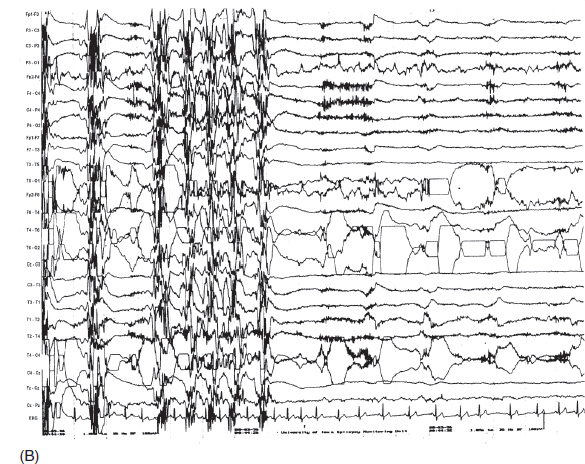
FIGURE 4.13 (A) A generalized ictal event in a 25-year-old man with a history of grand mal seizures since childhood. Note the sudden flattening of electroencephalography (EEG) activity at the onset, followed by beta activity peeking through the massive electromyogram (EMG) artifact during the tonic phase of seizure. (B) Toward the end of the seizure, ictal discharges changed to periodic spike-wave discharges, which were contaminated by muscle artifact (clonic phase). Afterward, there was postictal suppression of EEG activity.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
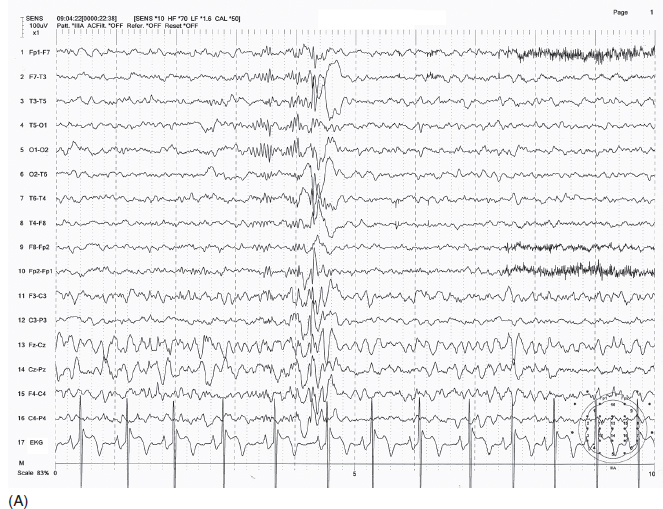
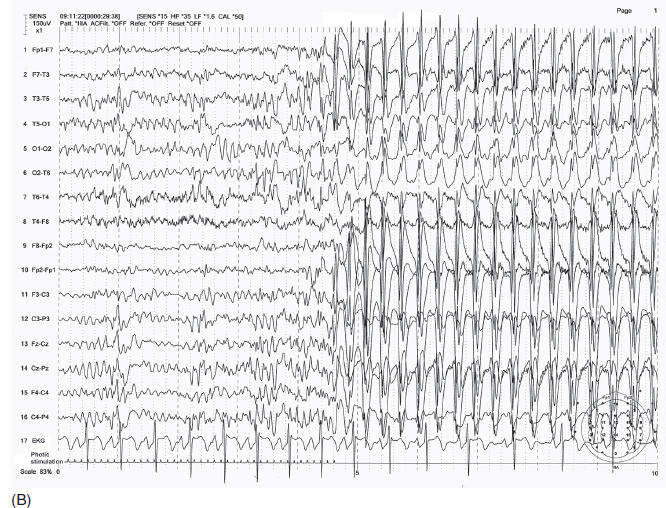
FIGURE 4.14 (A) An example of juvenile myoclonic epilepsy (JME) in a 14-year-old girl. Brief irregular (non-3 Hz) spike-wave discharges in sleep. (B) Photic stimulation–induced 3 Hz spike-wave bursts.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Hyperventilation may activate epileptiform activity in JME but less often as compared to absence seizures. About 30% to 40% of the patients also have photoparoxysmal seizures (Figure 4.14B) (39). Photosensitive epilepsy is three to four times more common in girls than in boys (39). In contrast to other types of epilepsy, epileptiform activity tends to decrease in sleep but markedly increases shortly after awakening. The ictal pattern is indistinguishable from interictal epileptiform activity in most cases but may have a greater number of polyspikes with higher amplitude.
Symptomatic Generalized, Multifocal Seizures or Syndromes
Lennox-Gastaut Syndrome
W.G. Lennox (40) and later H. Gastaut (41) described the clinical and electroencephalographic features of this disorder. LGS represents characteristic triads comprising (a) severe generalized seizures, (b) mental retardation, and (c) an EEG showing slow spike-and-wave (SSW) complexes. The SSW complexes consist of biphasic or triphasic sharp or spike waves followed by high-voltage (300–400 µV or greater) slow waves (Figure 4.15). Frequency of SSW complexes is between 1.5 and 2.5 Hz and is slower and often more irregular than the 3 Hz spike-wave complexes associated with idiopathic absence epilepsy. The bursts are usually bilaterally synchronous but may show shifting or persistent asymmetries. Asymmetric bursts may be associated with a unilateral lesion (42). In contrast to idiopathic absence epilepsy, which is usually associated with normal background activity, background activity in LGS is slow in more than 70% of the patients (43). Unlike the 3 Hz spike-wave discharges of absence seizures, both hyperventilation and photic stimulation are less effective in eliciting spike-wave discharges (44). In sleep, SSW complexes may become polyspike-wave discharges (44). Also paroxysmal fast activity is common during sleep (45). In addition to SSW, focal or multifocal epileptiform discharges may be seen in some patients (46).
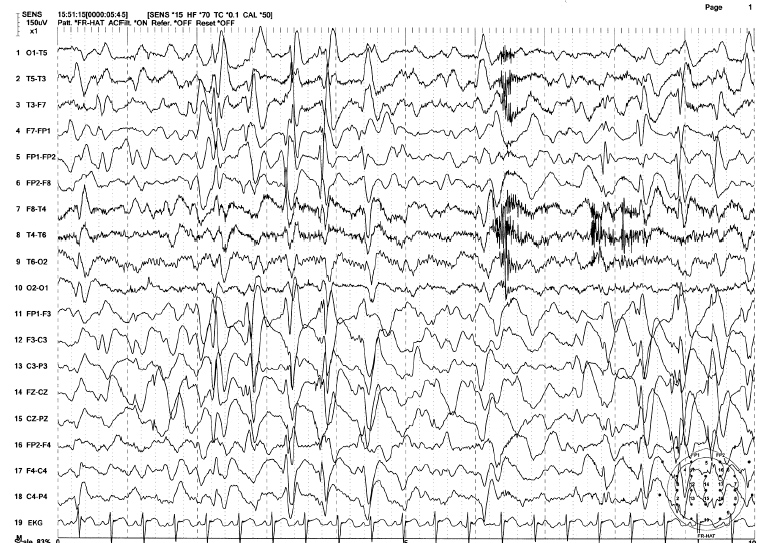
FIGURE 4.15 Generalized 2 Hz spike-wave bursts in an 18-year-old mentally disabled male with a history of intractable generalized tonic-clonic, tonic, and atypical absence seizures (Lennox-Gastaut syndrome). Note the high-voltage slow (2 Hz) spike-and-wave complexes associated with abundant irregular delta-theta activity in this awake EEG.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Median age of seizure onset in LGS is about 1 year and SSW complexes appear by 3 years. Greater than 75% have more than one type of seizure. The most common seizures are tonic seizure and atypical absence. Tonic seizures tend to appear at an earlier age and consist of sudden flexion of the hips, upper trunk, and neck, as well as arm abduction, elevation, or semiflexion. Because the seizure resembles infantile spasms, it can be considered a mature form of infantile spasms (47). Tonic seizures are associated with paroxysmal fast activity, often preceded by EEG flattening. The clinical distinction between typical and atypical absence seizures is not always clear but some features distinguish them. In atypical absence, impairment of consciousness is incomplete, rendering the transitions between normal activity and seizure activity unclear. Other symptoms such as eyelid or mouth myoclonus, changes in muscle tone, excessive salivation, and automatisms are common in atypical absence.
Ictal EEG change in atypical absence may be difficult to distinguish from the interictal pattern because both are represented by SSW complexes. The ictal pattern, however, tends to be more rhythmic, more widely distributed, and lasts longer than the interictal event (44). Other seizure types in LGS include clonic or tonic-clonic, atonic, myoclonic, and infantile spasms. The least common seizure is partial complex seizure.
INFANTILE SPASMS, SALAAM SPASMS, AND WEST SYNDROME Gibbs and Gibbs (48) first coined hypsarrhythmia (hyps means mountainous) to characterize the EEG pattern of “very high-voltage” (usually greater than 500 µV) irregular, asynchronous delta slow waves associated with multifocal spikes (Figure 4.16A). Spikes may be obscured by high-amplitude delta activity. The chaotic high-amplitude slow wave activity may be intermittently replaced by a relatively low-amplitude pattern (partial flattening) lasting 1 to 2 seconds. Because of exceedingly high-amplitude slow waves, waveforms are typically truncated in a recording using routine sensitivity (S = 7). Also, spikes are often hidden among large slow waves, and multifocal spikes are better visualized by using a shorter time constant (higher low-frequency filter) (Figure 4.16B). Typical hypsarrhythmia is common in younger infants, and over time the degree of abnormality tends to lessen to produce more organized activity with greater synchrony and symmetry and lower amplitude (49). The pattern may become modified, in which generalized sharp and slow-wave bursts become more synchronous within one hemisphere or between the two hemispheres (50). In cases of large focal lesions such as cysts or porencephaly, the hypsarrhythmic pattern may be unilateral (asymmetric hypsarrhythmia) or associated with persistent focal spikes or sharp waves (Figure 4.17). These variations may be classified as modified hypsarrhythmia. The hypsarrhythmic pattern is much more common in non-REM sleep than in the awake state or REM sleep (51). In non-REM sleep, bursts may become associated with longer attenuation periods. Hypsarrhythmia is commonly, but not always, associated with the clinical syndrome of infantile spasms or West syndrome (52). Infantile spasms may also be seen in patients with Aicardi syndrome with a distinct EEG pattern characterized by completely asynchronous burst suppression and multifocal spikes (Figure 4.18) (53). Seizures consist of brief flexion of the neck, trunk, and extremities. This sudden flexed motion is called a jackknife seizure or salaam attack. Seizures tend to occur in clusters shortly after awakening. The most common ictal EEG pattern associated with flexion spasms is sudden cessation of paroxysmal activity replaced by low-voltage fast activity or flattening of EEG activity, termed an electrodecremental seizure (Figure 4.19A–B). Other ictal patterns include frontal-dominant, high-amplitude rhythmic delta bursts or, less commonly, generalized spike- or sharp-wave complexes.
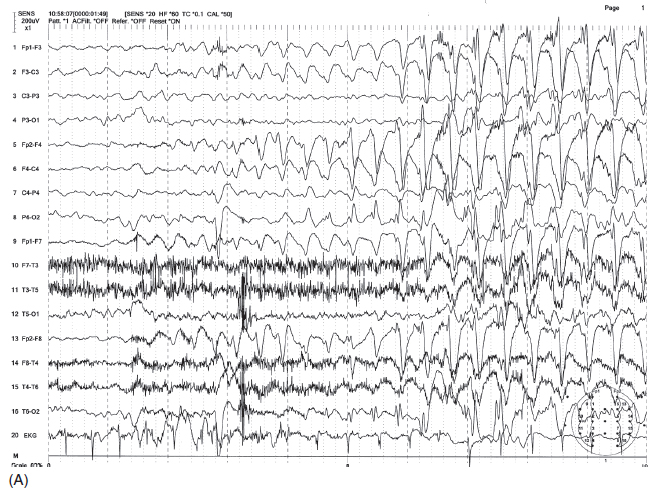
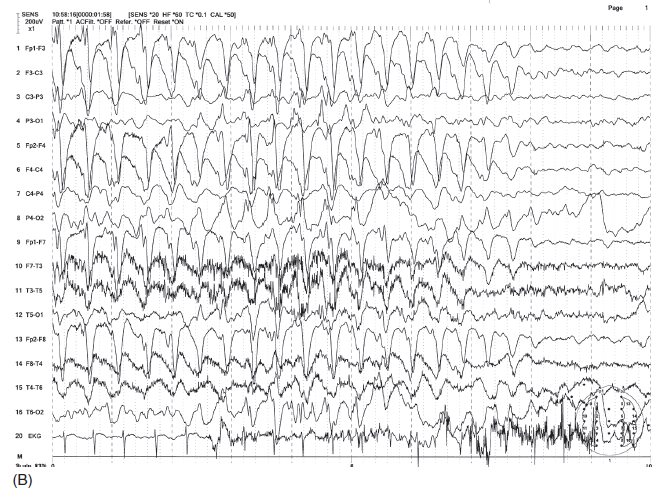
FIGURE 4.16 (A) Hypsarrhythmia in an 18-month-old microcephalic boy with infantile spasms. Note the high-amplitude irregular delta activity mixed with multifocal spikes and characteristic brief episodes of quiescence between bursts. (B) The evidence of multifocal and scattered spikes are better visualized by eliminating slow waves using a shorter time constant (0.03 seconds) or lower filter setting of 5 Hz (A and B are the same EEG samples).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
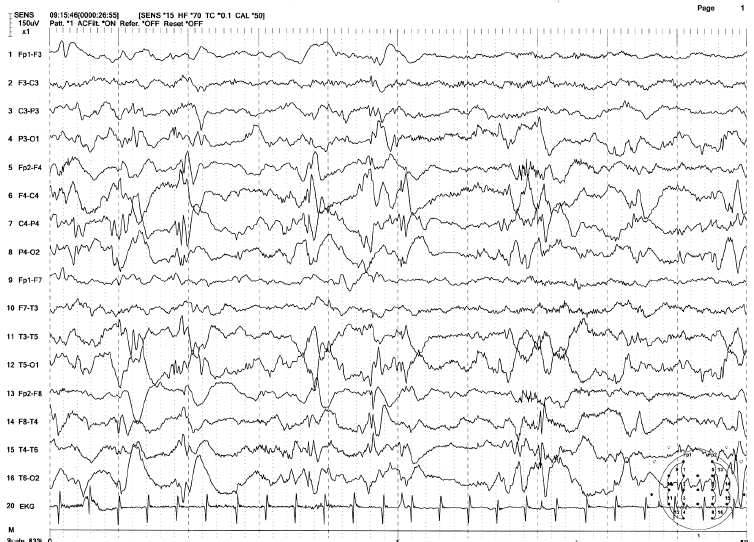
FIGURE 4.17 Modified hypsarrhythmia in an 8-month-old girl with a history of group B streptococcal meningitis at 2 weeks of age. The patient had frequent body jerks representing infantile spasms. Note the high-amplitude irregular spike-wave discharges from the right hemisphere, with brief flattening periods between bursts.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
The majority (>95%) of infantile spasms begin before the age of 1 year. Etiologies are diverse and include hereditary metabolic disorders, intrauterine infection, cerebral dysgenesis, tuberous sclerosis, hypoxic encephalopathy, etc. After adrenocorticotropic hormone (ACTH) or prednisone therapy, more than 60% of the patients improve dramatically with normalization of the EEG (54). Hypsarrhythmia disappears by 5 years; the EEG becomes normal in about half of the patients, while others continue to show various epileptiform discharges, focal, multifocal, or generalized including an SSW pattern (LGS). Normalization of the EEG does not necessarily indicate a normal neurologic state; nearly 90% of the patients remain disabled by epilepsy and other neurological deficits including severe mental impairment.
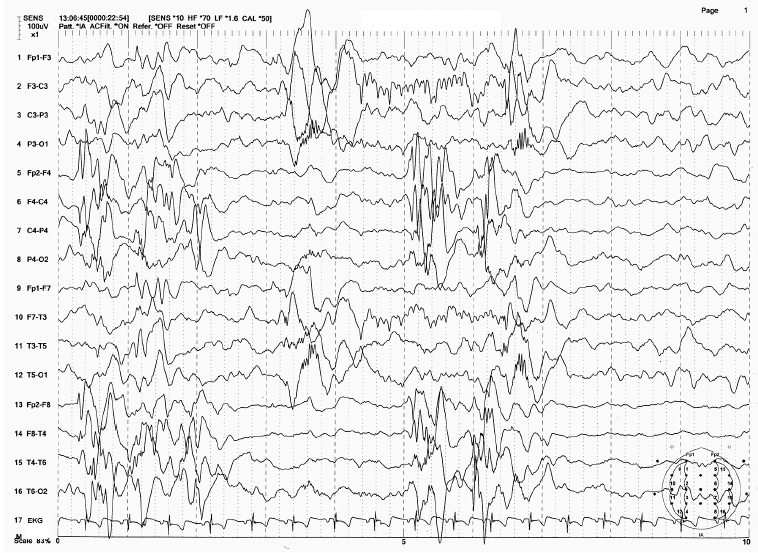
FIGURE 4.18 Electroencephalogram (EEG) of Aicardi syndrome in a 15-month-old girl with infantile spasms and hypsarrhythmic EEG. Note the gross asynchrony of EEG patterns between the two hemispheres.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Landau-Kleffner Syndrome
This syndrome, first described by Landau and Kleffner (55), is diagnosed based on characteristic clinical presentation and EEG abnormalities. Landau-Kleffner syndrome (LKS) affects 3- to 9-year-old children who were previously in good health. The first clinical sign is aphasia, which progressively worsens. Speech becomes progressively less intelligible and is eventually limited to only a few words (56). Hyperactivity and personality changes may appear as the aphasia worsens. About two-thirds of the patients have seizures of various types, including myoclonus, partial motor, akinetic/atonic, atypical absence, and generalized tonic-clonic convulsions (56).
The EEG in LKS is characterized by abundant epileptiform activity that is extremely variable in both location and volume; because of the characteristic deterioration of speech function, one may assume that the dominant hemisphere (left) is primarily affected. In some cases, the epileptiform activity indeed affects preferentially the left temporal region. But surprisingly, the majority of cases show variable patterns ranging from unifocal to multifocal and generalized spike waves. In the early stages of the illness, epileptiform activity may appear only in sleep. As the disease progresses, EEG abnormalities change considerably in terms of location, abundance, and pattern. Eventually, spikes and spike-wave discharges become more or less continuous, resulting in an appearance of “electrographic status epilepticus” in sleep (Figure 4.20A–B). This EEG feature is similar to that seen in the syndrome of continuous spikes and waves during slow-wave sleep (CSWS) (57). In fact, LKS and CSWS overlap in both clinical and electrographic features.
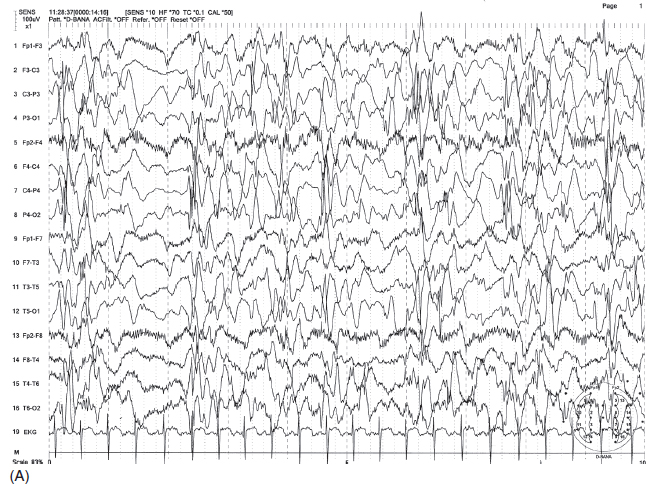
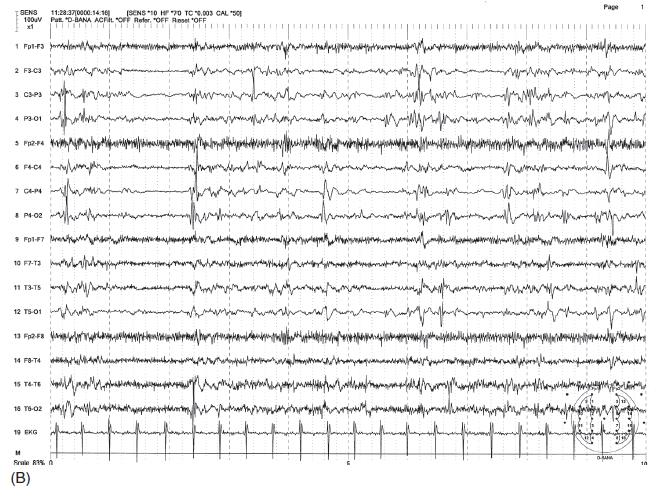
FIGURE 4.19 (A) Electrodecremental seizure associated with infantile spasms (ictal event) in a 2.5-year-old child with a history of hypoxic encephalopathy and severe developmental delay. Note the sudden flattening of electroencephalographic (EEG) activity accompanied by beta activity with concomitant increase of muscle tone artifact associated with body and arm jerks. (B) Interictal hypsarrhythmic pattern of this patient.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
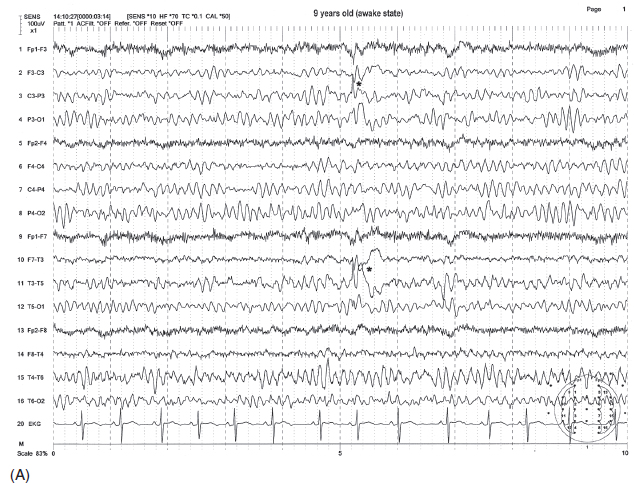
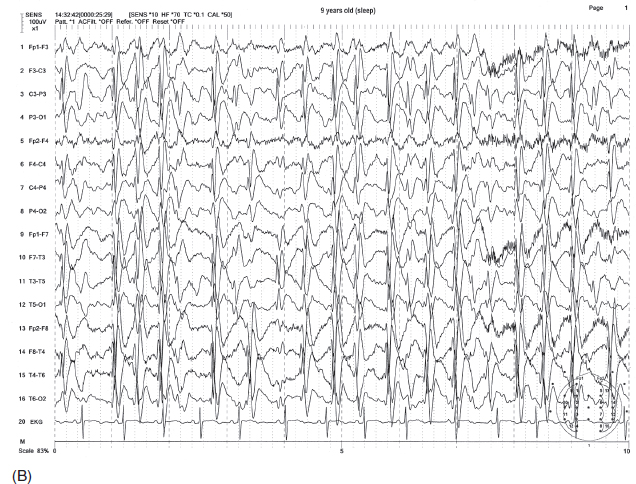
FIGURE 4.20 Dramatic electroencephalographic (EEG) change from awake (A) to asleep (B) state in a 9-year-old boy with the diagnosis of Landau-Kleffner syndrome. Note the sporadic sharp wave discharges from the left hemisphere (indicated by the * in A) in the awake state and the more or less continuous generalized spike-wave bursts becoming electrographic status epilepticus in sleep (B).
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Despite the severe degree of clinical as well as EEG abnormalities, many patients recover, with normal EEG and seizure remission, but some degree of language dysfunction may persist (56,58).
Continuous Spike and Wave During Slow-Wave Sleep
CSWS was first described by Patry (59). The syndrome is characterized by continuous spike and wave activity during non-REM sleep (56,60) and is sometimes referred to as epilepsy with electrical status epilepticus during slow sleep (ESES) (Figure 4.21A–B) (60). The age of onset ranges from 1 to 12 years, but most occur around 8 years. Two-thirds of the patients are neurologically normal before onset. In time, most patients have frequent seizures (generalized tonic-clonic, atypical absence, and atonic) and have a significant decline in IQ with deterioration in language, impaired memory, reduced attention span, and behavioral changes with aggression or psychosis (56,60).
Epileptiform activity consists of generalized SSWs (1.5–2.5 Hz) as well as focal or multifocal spikes, which are sporadic in the waking state. In sleep, spike-wave bursts become nearly continuous (CSWS pattern), occupying more than 85% of the total non-REM sleep time (60). The CSWS pattern persists for 1 to several years. Similar to LKS, the EEG then tends to normalize and seizures remit spontaneously in most patients. However, recovery of neurological deficit and behavior is often incomplete and about one-half of the patients remain profoundly impaired (56,60).
Subacute Sclerosing Panencephalitis
Subacute sclerosing panencephalitis (SSPE) is a sequel of the measles infection and has become extremely rare because of mandatory immunization. The periodic burst activity in SSPE consists of high-amplitude slow waves mixed with sharp waves, occasionally including spike discharges, maximum in the frontocentral region (Figure 4.22A). The bursts repeat at 4- to 15-second intervals (61) and tend to be more prominent during the awake state. Because of the relatively slow periodicity, a slower sweep speed (20–30 sec/page) makes it easier to rate the repetition (Figure 4.22B). Patients with SSPE often have myoclonus coinciding with the EEG bursts. Background activity becomes slow as the disease progresses. The origin of the periodic discharges has been debated as either thalamic (62) or cortical (63).
Progressive Myoclonic Epilepsy
There are several clinical entities that account for progressive myoclonic epilepsies. They are myoclonic epilepsies with ragged red fibers (MERRF syndrome) (64), Lafora disease (65), Unverricht-Lundborg disease (Baltic myoclonic epilepsy) (66), neuronal ceroid lipofuscinosis (Batten disease) (67), and sialidosis (cherry-red spot myoclonus syndrome) (68). These syndromes are characterized by myoclonic seizures, progressive ataxia, and dementia secondary to degenerative central nervous system (CNS) disease with metabolic derangement. In all forms of progressive myoclonic epilepsies, epileptiform activity consists of generalized spike-waves, polyspike-waves, and multifocal spikes. Background activity becomes progressively slower as the disease process progresses. Photoparoxysmal responses are common in patients with Lafora disease, Unverricht-Lundborg disease, and Batten disease. In Batten disease, single or low-frequency intermittent photic stimulation characteristically produces prominent spike discharges at occipital electrodes with a one-to-one relationship with the light flash (67) (Figure 4.23).
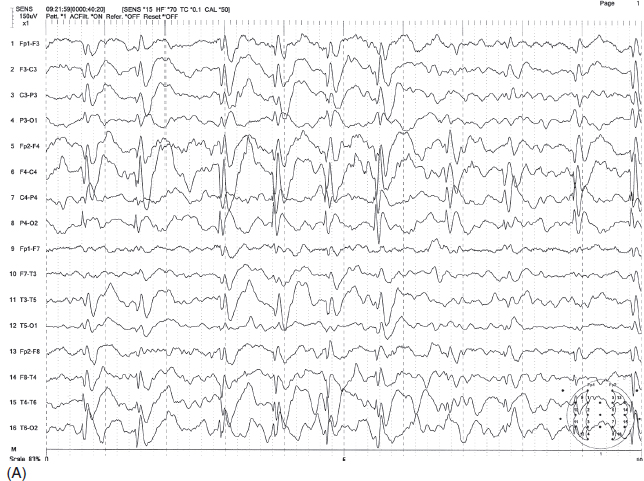
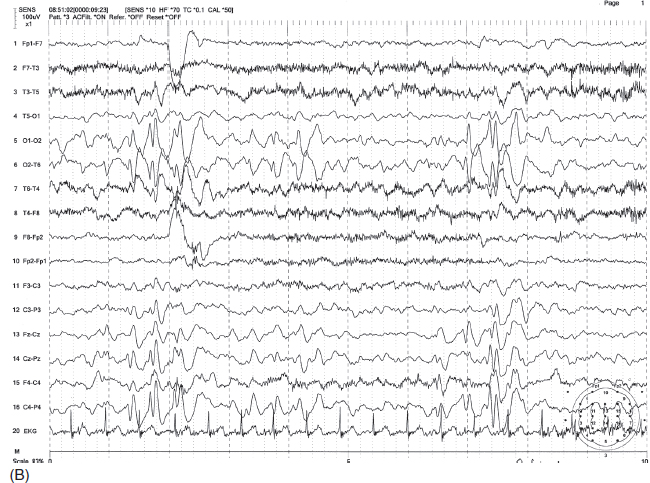
FIGURE 4.21 (A) Continuous spike-wave discharges during slow-wave sleep (CSWS) in a 7-year-old with a history of generalized tonic-clonic seizures mostly during sleep and atypical absence associated with drop attacks. Note the continuous right-greater-than-left parasagittal-dominant spike-wave bursts in sleep. (B) These discharges are much less prominent in the awake state.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
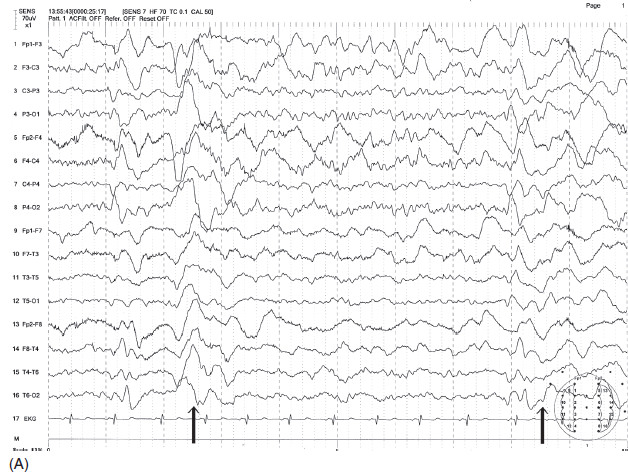
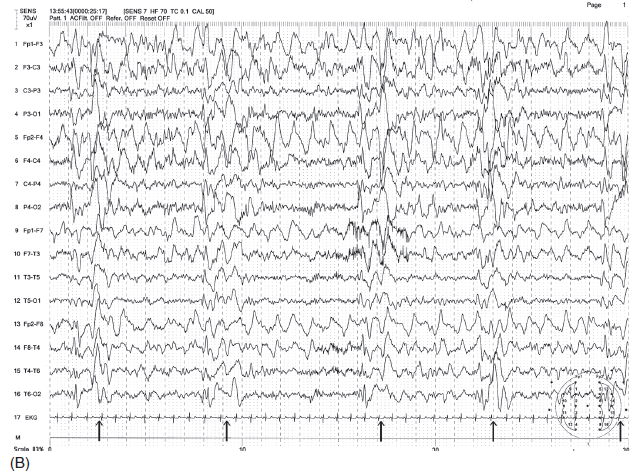
FIGURE 4.22 (A) Generalized periodic discharges in a 15-year-old boy with a diagnosis of subacute sclerosing panencephalitis (SSPE). Note the two bursts of irregular delta waves occurring in about 5-second intervals (indicated by arrows). (B) Because of slow recurrence, the periodicity was not clear with the routine sweep speed of 10 seconds/page, but this became more evident (indicated by arrows) with a slower sweep speed of 30 seconds/page. (The first 10 seconds in Figure B are actually represented by Figure A at a faster sweep of 10 seconds/page.)
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Rasmussen Encephalitis
These seizures consist of refractory focal motor seizures or prolonged episodes of epilepsia partialis continua, associated with progressive hemiatrophy with contralateral hemiparesis (69,70). EEG shows more or less continuous ictal spike discharges associated with focal motor seizures or epilepsia partialis continua. In some cases, no ictal discharges may be detected despite obvious clinical seizures if the seizure discharges arise from a deep source or limited cortical zone. The diagnostic criteria were outlined by a recent European conference based on clinical, EEG, and MRI findings. This consists of (a) clinical focal seizures with or without epilepsia partialis continua and unilateral cortical deficit, (b) EEG showing unilateral slowing with or without epileptiform activity and unilateral seizure onset, and (c) MRI demonstrating unihemispheric cortical atrophy and at least one either gray or white matter hyperintensity or atrophy of the caudate (70). The treatments include autoimmune treatment, limited cortical excisions, lobectomies, or hemispherectomies, but hemispherectomy seems to be the most effective treatment (70).
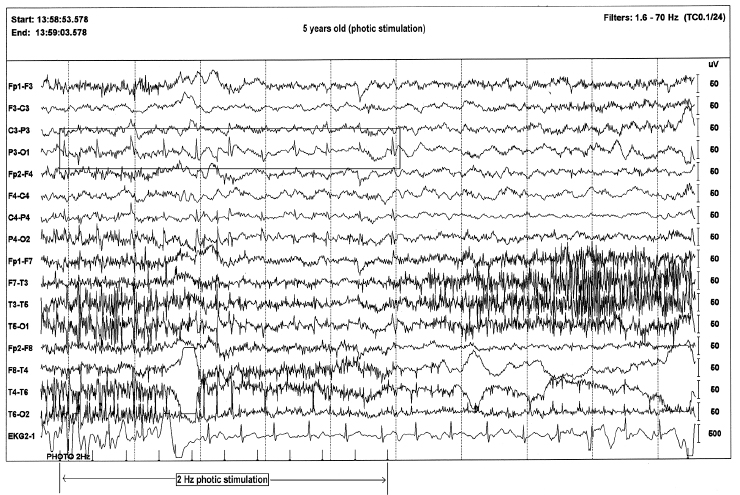
FIGURE 4.23 Photic stimulation–induced spikes in a 5-year-old boy with a diagnosis of Batten disease. The time-locked spikes with slow photic stimulation is characteristic of this diagnosis.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
Clinical manifestations of this epilepsy include sudden awakening with violent movement, dystonic or tonic posturing, and hyperactive behavior. The event resembles night terror or nocturnal paroxysmal dystonia. EEG may be normal or may show frontal slowing or spikes. The ictal EEG may show rhythmic frontal-dominant discharges, but more importantly there may be no overt EEG changes associated with the event (71).
Multifocal Independent Spikes Syndrome
This is defined by three or more independent spike foci arising from various locations (72) (Figure 4.24) and commonly appears between age 4 and 7 years. This could occur after hypsarrhythmia or LGS. EEG is usually associated with slow background activity and multifocal spikes in the awake state which may become more frequent and more synchronized in sleep. The clinical seizures often appear as tonic spasms, but generalized tonic-clonic seizures or focal seizures are possible (73). The EEG, especially with normal background activity, may show dramatic improvement in time with normal neurologic finding.
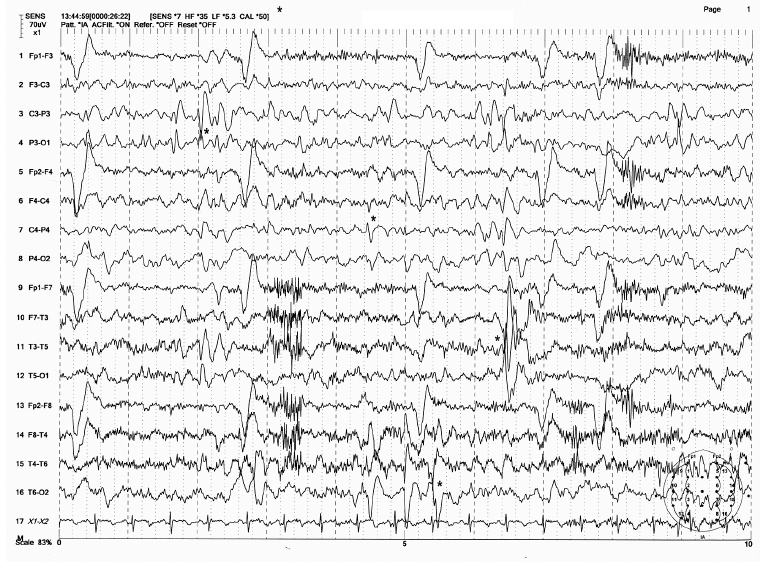
FIGURE 4.24 Multifocal sharp and spike discharges in a 4-year-old boy with a history of partial complex seizures and generalized tonic-clonic convulsions. There are at least four spike foci appearing as single or repetitive transients from P3, C4, T3, and T6 electrodes (shown by * marks) independently.
Source: From Ref. (74). Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010; with permission.
REFERENCES
1. Gotman J, Marciani MG. Electroencephalographic spiking activity, drug levels, and seizure occurrence in epileptic patients. Ann Neurol. 1985;17:597–603.
2. Eeg-Olofsson O, Petersen I, Sellden U. The development of electroencephalogram in normal children from the age of 1 though 15 years. Paroxysmal activity. Neuropaediatric. 1971;2:375–404.
3. Cavazzuti GB, Cappella L, Nalin A. Longitudinal study of epileptiform EEG patterns in normal children. Epilepsia. 1980;21:43–55.
4. Bennet DR. Spike wave complexes in “normal” flying personnel. Aerosp Med.1967;38:1276–1282.
5. Kellaway P. The incidence, significance, and natural history of spike foci in children. In Henary CE, ed. Current Clinical Neurophysiology: Update on EEG and Evoked Potentials. Amsterdam: Elsevier;1981:151–175.
6. Ehle A, Co S, Jones MG, Clinical correlates of midline spike. An analysis of 21 patients. Arch Neurol. 1981;38:355–357.
7. Marsan CA, Zivin LS. Factors related to the occurrence of typical paroxysmal abnormalities in the EEG records of epileptic patients. Epilepsia. 1970;11:361–381.
8. Salinsky M, Kanter R, Dasheiff RM. Effectiveness of multiple EEGs in supporting the diagnosis of epilepsy: an operation curve. Epilepsia. 1989;28:331–334.
9. Berg AT, Berkovic SF, Brodie MJ, et al. Revised termimology and concept for seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, Epilepsia. 2010;51:676–685.
10. Blom S, Heijbel J. Benign epilepsy of children with centro-temporal EEG foci: Discharge rate during sleep. Epilepsia. 1975;16:133–140.
11. Loiseau P, Ducke B, Cordova S, et al. Prognosis of benign childhood epilepsy with centro temporal spikes: a follow up study of 168 patients. Epilepsia. 1988;29:229–235.
12. Gregory DL, Wong PK. Topographic analysis of the centrotemporal discharges in benign rolandic epilepsy of childhood. Epilepsia. 1984;25:705–711.
13. Legarda S, Jayakar P, Duchowny M, et al. Benign rolandic epilepsy: high central and low central subgroups. Epilepsia. 1994;35:1125–1129.
14. Gastaut H. A new type of epilepsy: benign partial epilepsy of childhood with occipital spike-waves. Clin Electroencephalogr. 1982;13:13–22.
15. Panayiotopoulos CP. Benign childhood epileptic syndromes with occipital spikes: new classification proposed by the International League Against Epilepsy. J Child Neurol. 2000;15:548–552.
16. Gastaut H. Zifken BG. Benign epilepsy of childhood with occipital spike and wave complexes. In: Andermann F, Lugaresi E, eds. Migraine and Epilepsy. Boston, MA: Butterworth;1987:47–81.
17. Panayiotopoulos CP. Early-onset benign childhood occipital seizure susceptibility syndrome; syndrome to recognize Epilepsia. 1999;40:621–630.
18. Ferrie CD, Beaumanoir A, Guerrini R, et al. Early-onset benign occipital seizure susceptibility syndrome. Epilepsia. 1997;38:285–293.
19. Kellaway P. The incidence, significance and natural history of spike foci in children: In: Henry CE, ed. Current clinical neurophysiology: update on EEG and evoked potentials. Amsterdam: Elsevier;1981:151–175.
20. Guerrini R, Dravet C, Genton P, et al. Idiopathic photosensitive occipital lobe epilepsy. Epilepsia. 1995;36:883–891.
21. Cooper GW, Lee SI. Reactive occipital epileptiform activity: is it benign? Epilepsia. 1991;32: 63–68.
22. Gibbs FA, Davis H, Lennox WG. The electroencephalogram in epilepsy and in conditions of impaired consciousness. Arch Neurol Psychiatry. 1935;34:1133–1148.
23. Rodin E, Ancheta O. Cerebral electrical fields during petit mal absences. Electroencepharogr Clin Neurophysiol. 1987;66:457–466.
24. Sato S, Dreifuss FE, Perry JK, et al. Long-term follow-up of absence seizures. Neurology. 1983;33:1590–1595.
25. Wolf P. Juvenile absence epilepsy In: Roger J, Bureau M, Dravet C, et al, eds. Epileptic syndrome in infancy, childhood and adolescence. London: John Libbey; 1992:307–312.
26. Sato S, Dreifuss FE, Penry JK. The effect of sleep on spike-wave discharge in absence seizures. Neurology. 1973;23:1335–1345.
27. Sato, White BG, Penry JK, et al. Valproic acid versus ethosuximide in the treatment of absence seizures. Neurology. 1982;32:157–163.
29. Holmes GL, McKeever M, Adamson M. Absence seizures in children: clinical and electroencephalographic features. Ann Neurol. 1987;21:268–273.
30. Browne TR, Penry JK, Porter RJ, et al. Responsiveness before, during and after spike-wave paroxysms. Neurology. 1974;24:659–665.
31. Penry JK, Dreifuss FE. A study of automatisms associated with the absence of petit mal. Epilepsia. 1969;10:417–418.
32. Camfield CS, Camfields PR, Sadler M, et al. Paroxysmal eyelid movements: a confusing feature of generalized photosensitive epilepsy. Neurology. 2004;63:40–42.
33. Loiseau P. Childhood absence epilepsies. In: Roger J, Dravert C, Bureau M, et al, eds. Epileptic Syndrome in Infancy and Adolescence. London: John Libbey;1985:106–120.
34. Sato S, Dreifuss FE, Perry JK, et al. Long-term follow-up of absence seizures. Neurology. 1983;33:1590–1595.
35. Wolf P, Juvenile absence epilepsy. In: Roger J, Dravert C, Bureau M, et al, eds. Epileptic Syndrome in Infancy and Adolescence. London: John Libbey;1992:307–312.
36. Niedermeyer E. Generalized seizure discharges and possible precipitating mechanisms. Epilepsia. 1966;7:23–29.
37. Janz D, Christian W. Impulsive-petit mal. Dtsch Z Nervenheik. 1957;176:346–386.
38. Delgado – Escueta AV, Enrile-Bascal F. Juvenile myoclonic epilepsy of Janz. Neurology. 1984;34:285–294.
39. Wolf P, Groosses R. Relation of photosensitivity to epileptic syndrome. J Neurol Neurosurg Psychiatry. 1986;49:1386–1391.
40. Lennox WG. Epilepsy and related disorders. Boston, MA: Little Brown; 1960
41. Gastaut H, Broughton R. Epileptic Seizures. Springfield, IL: Charles C Thomas; 1972.
42. Gastaut H, Roger J, Soulayrol R, et al. Epileptic encephalopathy of children with diffuse slow spikes and waves (alias “petit mal variant”) or Lennox syndrome. Ann Pediatr. 1966;13:489–499.
43. Blume WT. Lennox-Gastaut syndrome. In: Luders H, Lesser RP, eds. Epilepsy: electroclinical syndromes. London: Springer – Verlag;1987:73–92.
44. Markand ON. Slow spike-wave activity in EEG and associated clinical feature: often called “Lennox” or “Lennox-Gastaut” syndrome. Neurology. 1977;27:746–757.
45. Beaumanoir A. The Lennox – Gastaut syndrome: a personal study. Electroencepharogr Clin Neurophysiol Suppl. 1982;35:85–99.
46. Blume WT, David RB, Gomez MR. Generalized sharp and slow wave complexes. Associated clinical features and long-term follow up. Brain. 1973;96:289–306.
47. Egli M, Mothersill I, O’Kane N, et al. The axial spasm—the predominant type of drop seizure in patients with secondary generalized epilepsy. Epilepsia. 1985;26:401–415.
48. Gibbs FA, Gibbs JD. Atlas of Encephalography. Cambridge, MA: Addison-Wesley; 1952
49. Kellaway P, Frost JD Jr, Hrachovy RA. Infantile spasms. In: Morselli PD, Pippinger KF, Penry JK, eds. Antiepileptic drug therapy in pediatrics. New York, NY: Raven Press; 1983:115–136.
50. Hrachovy RA, Frost JD Jr, Kellaway P. Hypsarrythmia: variations on theme. Epilepsia. 1984;25:317–325.
51. Watanabe K, Negoro T, Aso K et al. Reappraisal of interictal electroencephalogram in infantile spasms. Epilepsia. 1993;34: 679–685.
52. West WJ. On a particular form of infantile convulsions. Lancet. 1841;1:724–725.
53. Fariello RG, Chen RW, Doro JM et al. EEG recognition of Aicardi’s syndrome. Arch Neurol. 1977;34:563–566.
54. Hrachovy RA, Frost JD Jr, Kellaway P, Zion TE. Double–blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103:641–645.
55. Landau WM, Kleffner FR. Syndrome of acquired aphasia with convulsive disorder in children. Neurology. 1957;7:523–530.
56. Beaumanoir A. EEG data: In Beaumanoir A, Bureau M, Deonnat T, et al, eds. Continuous spike and waves during slow sleep electrical status epileptics during slow sleep. London: John Libbey; 1995:217–223.
58. Deonna T, Peter C, Ziegler AL. Adult follow-up of the acquired aphasia – epilepsy syndrome in childhood. Report of 7 cases. Neuropediatrics. 1989;20:132–138.
59. Patry G, Lyagoubi S, Tassinari CA. Subclinical “electrical status epilepticus” induced by sleep in children. A clinical and electroencephalographic study of six cases. Arch Neurol. 1971;24:242–252.
60. Tassinari CA, Bureau M, Dravet C, et al. Epilepsy with continuous spikes and waves during slow wave sleep—otherwise described as ESES (epilepsy with electrical status epilepticus during slow sleep). In Roger J, Bureau M, Dravet C, et al, eds. Epileptic syndrome in infancy, childhood and adolescence. London: John Libbey; 1992:245–256.
61. Rabending G, Radermecker FJ. Subacute sclerosing panencephalitis (SSPE). In Remmon A (ed-in-chief). Handbook of Electroencephalopathy and Clinical Neurophysiology. Vol 15A. Amsterdam: Elsevier;1977:28–35.
62. Radermecker J, Poser CM. The significance of repetitive paroxysmal electroencephalographic pattern. Their specificity in subacute sclerosing leukoencephalitis. World Neurol. 1960;1:422–433.
63. Storm van Leeuwen W. Electroencephalographical and neurophysiological aspects of subacute sclerosing leuco-encephalitis. Psychiatr Neurol Neurochir. 1964;67:312–322.
64. Ohtsuka Y, Amano R, Oka E, et al. Myoclonus epilepsy with ragged-red fibers: a clinical and electrophysiological study on two siblings cases. J Child Neurol. 1993;8:366–372.
65. Reese K, Toro C, Malow B, et al. Progression of the EEG in Lafora-body disease. Am J EEG Technol. 1993;33:229–235.
66. Roger J, Genton P, Bureau M, et al. Progressive myoclonus epilepsies in childhood and adolescence. In Roger J, Bureau M, Dreifuss FF, et al, eds. Epileptic syndromes in infancy, childhood and adolescence. 2nd ed. London: Libbey; 1992:381–400.
67. Pampiglione G, Harden A. So-called neuronal ceroid lipofuscinosis. Neurophysiological studies in 60 children. J Neurol Neurosurg Psychiatry. 1977;40:323–330.
68. Engel J Jr, Rapin I, Giblin DR. Electroencephalographical studies in two patients with cherry red spot-myoclonus syndrome. Epilepsia. 1977;18:73–87.
69. Rasmussen T, Olszewski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8:435–445.
70. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, Diagnosis, and treatment of Rasmussen encephalitis. A European consensus statement. Brain. 2005;128:454–471.
71. Raju P, Sarco DP, Poduri A, et al. Oxcarbazepine in children with nocturnal frontal lobe epilepsy. Pediat Neurol. 2007;37:345–349.
72. Blume WT. Clinical and electroencephalographic correlates of the multiple independent spike foci in children. Ann Neurol. 1978;4:541–547.
73. Yamatogi Y, Ohtahara S. Multiple independent spike foci with epilepsy, with special reference to a new epileptic syndrome of “severe epilepsy with multiple independent spike foci in children”. Epilepsy Res. 2006;70(S1):541–547.
74. Yamada T, Meng E. Practical Guide for Clinical Neurophysiologic Testing: EEG. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2010.
Related posts:
 Clinical Neurophysiology: Future Role in Pediatric Neurologic Disorders
Pediatric Febrile Seizures
The Evaluation of Pediatric Sleep Disorders
EEG Monitoring in Neonatal Epilepsies
Evoked Potentials in Adolescent Idiopathic Scoliosis: Intraoperative Neurophysiological Monitoring
EMG in Pediatric Brachial Plexopathy
Clinical Neurophysiology: Future Role in Pediatric Neurologic Disorders
Pediatric Febrile Seizures
The Evaluation of Pediatric Sleep Disorders
EEG Monitoring in Neonatal Epilepsies
Evoked Potentials in Adolescent Idiopathic Scoliosis: Intraoperative Neurophysiological Monitoring
EMG in Pediatric Brachial Plexopathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


