9 Endocrinology Skull-based endocrinology focuses on the management of pituitary/hypothalamic disruption by sellar and suprasellar lesions. There is a broad differential for lesions in the sellar and suprasellar areas (see Chapter 21, Table 21.1 on page 498). The mnemonic CAM-LIGHTS can be used to remember these lesions: Cyst, craniopharyngioma, chordoma, carcinoma, Adenoma, abscess, arteriovenous fistulas, Meningioma, metastasis, Lymphoma, Infiltrative, Germ cell tumor, Hypophysitis, Histiocytosis X, Tuberculosis, Sarcoidosis. • All lesions can present with symptoms and signs of mass effect: headache, decreased visual acuity, relative afferent pupillary defect, bitemporal hemianopsia, pale optic disk, diplopia, ptosis, and facial numbness. A lactotroph tumor of the anterior lobe of the pituitary produces prolactin, resulting in the clinical presentation of galactorrhea, infertility, and oligo- or amenorrhea in women. • The majority of prolactinomas are larger in men than in women.1 • Men usually present later than women, and present with symptoms related to mass effect, such as headache and visual field defects. • Men may also present with symptoms of hypogonadotropic hypogonadism, such as infertility, decreased libido, and erectile dysfunction. Men may also present with symptoms of hypopituitarism.2 Prolactinoma accounts for approximately 40% of all pituitary tumors, with a prevalence variously noted to be 6 to 10 per 100,000 or 50 per 100,000.3,4 • Monoclonal expansion of a single cell that has likely undergone somatic mutation. • Pituitary tumor transforming gene (PTTG) is overexpressed,5 but it is expressed at higher levels in other pituitary tumors.6 • Familial and genetic considerations: Rule out other causes of elevated prolactin (Table 9.1). Table 9.1 Causes of Hyperprolactinemia8, 9
 Functional and Nonfunctional Pituitary Adenomas and Other Sellar Lesions
Functional and Nonfunctional Pituitary Adenomas and Other Sellar Lesions
Prolactinoma
Epidemiology
Pathophysiology
 Multiple endocrine neoplasia-1 (MEN1): mutation in the MEN1 gene, 40% of cases have a pituitary adenoma, most commonly a prolactinoma (other clinical manifestations include parathyroid adenomas, pancreatic islet cell/gastrointestinal adenomas)
Multiple endocrine neoplasia-1 (MEN1): mutation in the MEN1 gene, 40% of cases have a pituitary adenoma, most commonly a prolactinoma (other clinical manifestations include parathyroid adenomas, pancreatic islet cell/gastrointestinal adenomas)
 Carney’s complex: mutation in the PRKAR1A gene; presents with lentigines, myxomas, Schwann cell tumors, adrenal hyperplasia, and pituitary abnormalities (frequent hypersecretion of prolactin)
Carney’s complex: mutation in the PRKAR1A gene; presents with lentigines, myxomas, Schwann cell tumors, adrenal hyperplasia, and pituitary abnormalities (frequent hypersecretion of prolactin)
 Familial isolated pituitary adenomas: 15% have mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene, 40% are prolactinomas7
Familial isolated pituitary adenomas: 15% have mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene, 40% are prolactinomas7
Differential Diagnosis
Physiological causes | Pregnancy, lactation, stress, coitus, sleep, exercise, mammary stimulation |
Pathological sellar causes | Pituitary: prolactinoma, pituitary adenomas with mixed secretion of prolactin/growth hormone, surgery, trauma, hypophysitis Pituitary stalk damage: nonpituitary tumors (craniopharyngioma, germinoma, metastasis, meningioma), pituitary tumors other than prolactinoma, Rathke’s cyst, granulomas, infiltrative lesions, cranial irradiation |
Pharmacological causes | Antipsychotics especially neuroleptics, clomipramine, desipramine, amitriptyline, fluoxetine, metoclopramide, cimetidine, ranitidine, methyldopa, verapamil, labetalol, phenytoin, codeine, morphine, estrogens, protease inhibitors |
Other | Primary hypothyroidism, chest wall injury, chronic renal failure, cirrhosis, epileptic seizures, polycystic ovarian syndrome, idiopathic hyperprolactinemia, macroprolactinemia, genetic |
Diagnosis
• Measure prolactin level and request magnetic resonance imaging (MRI) of the sella.
• Levels generally correlate with tumor size; prolactin is usually > 200–250 μg/L (> 200–250 ng/mL) in macroprolactinomas.
• Rule out other causes of hyperprolactinemia (Table 9.1).
• Macroprolactinemia:
 Most prolactin is monomeric.
Most prolactin is monomeric.
 Macroprolactinemia occurs when there is a preponderance of a covalently bound polymeric form of inactive prolactin, which remains reactive to varying degrees in prolactin immunoassays.10
Macroprolactinemia occurs when there is a preponderance of a covalently bound polymeric form of inactive prolactin, which remains reactive to varying degrees in prolactin immunoassays.10
 Normal biological activity; be suspicious when typical symptoms are absent.
Normal biological activity; be suspicious when typical symptoms are absent.
 Order a macroprolactin level when investigating asymptomatic hyperprolactinemia.
Order a macroprolactin level when investigating asymptomatic hyperprolactinemia.
• Hook effect: assay artifact where a very high prolactin level saturates both the capture and signal antibodies, resulting in artifactually low prolactin results.11,12 If the Hook effect is suspected, the assay should be repeated after a serial serum sample dilution.
Medical Treatment
• Dopamine agonists
 Bromocriptine: Starting adult dose is 1.25 to 2.5 mg every night at bedtime, which can be increased by 2.5 mg as tolerated every 2 to 7 days until an optimal response is obtained. Maximum dose is 15 mg per day. Side effects include dizziness, headache, fatigue, nausea, and rhinitis.
Bromocriptine: Starting adult dose is 1.25 to 2.5 mg every night at bedtime, which can be increased by 2.5 mg as tolerated every 2 to 7 days until an optimal response is obtained. Maximum dose is 15 mg per day. Side effects include dizziness, headache, fatigue, nausea, and rhinitis.
 Cabergoline: Starting adult dose is 0.5 mg weekly or 0.25 mg twice a week, which can be increased by 0.5 mg every 4 weeks until an optimal response is obtained. Maximum dose is 2 mg per week. Higher doses have been used off-label in studies. Side effects include headache, dizziness, and nausea.
Cabergoline: Starting adult dose is 0.5 mg weekly or 0.25 mg twice a week, which can be increased by 0.5 mg every 4 weeks until an optimal response is obtained. Maximum dose is 2 mg per week. Higher doses have been used off-label in studies. Side effects include headache, dizziness, and nausea.
 Quinagolide: Starting adult dose is 0.025 mg per day for 3 days followed by 0.05 mg per day for 3 days, and then 0.075 mg per day. Further titration can be done at monthly intervals. The maximum dose is 0.9 mg per day. Side effects include dizziness, fatigue, headache, nausea, and vomiting.
Quinagolide: Starting adult dose is 0.025 mg per day for 3 days followed by 0.05 mg per day for 3 days, and then 0.075 mg per day. Further titration can be done at monthly intervals. The maximum dose is 0.9 mg per day. Side effects include dizziness, fatigue, headache, nausea, and vomiting.
 Treat patients who are symptomatic to restore gonadal function, decrease prolactin levels, and decrease tumor size.8
Treat patients who are symptomatic to restore gonadal function, decrease prolactin levels, and decrease tumor size.8
 First-line treatment is a dopamine agonist; cabergoline is more effective and better tolerated than bromocriptine.13
First-line treatment is a dopamine agonist; cabergoline is more effective and better tolerated than bromocriptine.13
• In a placebo-controlled study, cabergoline in microprolactinomas, idiopathic hyperprolactinemia and empty sella resulted in normalization of prolactin in 95% of patients on 1 mg twice a week. Menses was restored in 82% of women with amenorrhea.14
• In a prospective study with macroprolactinomas, normal prolactin levels were achieved within 6 months in 81% of patients receiving cabergoline, and 92% had significant tumor shrinkage.15
• Visual fields normalized in 33% of patients and improved in 56% of patients in one series.16
• Results are similar to the impact of surgery on visual fields for giant nonfunctional tumors in another series17; of those who had visual field defects preoperatively, after surgery, 8.6% remained blind, 28% regained normal sight, and 67% had variable improvement.
• If the patient is symptomatic and does not achieve normalization of prolactin level or tumor shrinkage, increase the medical therapy to the maximum tolerated doses prior to considering surgery. Maximum dose for cabergoline is 2 mg/week. Higher doses have been used off-label in studies. Monitor with two-dimensional (2D) echocardiogram for valvular regurgitation if on high-dose cabergoline (> 2 mg a week).8
• A cerebrospinal fluid (CSF) leak can occur if a defect is revealed in the sellar floor after tumor shrinkage with a dopamine agonist.
Withdrawal of dopamine agonist therapy:
• Can consider withdrawal of therapy in patients who have been treated with a dopamine agonist for 2 years who no longer have an elevated prolactin and who have had significant tumor reduction as demonstrated on MRI.8,18
• Factors predicting remission:
 Nadir prolactin level during treatment ≤ 5.4 µg/L (≤ 5.4 ng/mL) and nadir maximal tumor diameter ≤ 3.1 mm19
Nadir prolactin level during treatment ≤ 5.4 µg/L (≤ 5.4 ng/mL) and nadir maximal tumor diameter ≤ 3.1 mm19
 Size of tumor remnant prior to withdrawal of therapy: 18% increased recurrence risk for each millimeter20
Size of tumor remnant prior to withdrawal of therapy: 18% increased recurrence risk for each millimeter20
 Normalization of MRI and longer duration of treatment21
Normalization of MRI and longer duration of treatment21
• Rates of recurrence after withdrawal18,19,22–24:
 Idiopathic hyperprolactinemia 24–68%
Idiopathic hyperprolactinemia 24–68%
 Microadenomas 31–79%
Microadenomas 31–79%
 Macroadenomas 36–93%
Macroadenomas 36–93%
• Hyperprolactinemia is most likely to recur within the first year after withdrawal of dopamine agonists.20,24
• Follow prolactin every 3 months for 1 year, then annually; order an MRI if prolactin increases above the normal range.8
• Ongoing surveillance for increase in size of tumor is needed.
Other medical treatment options:
 Can also consider an oral contraceptive pill in pre-menopausal women with amenorrhea and microprolactinoma.
Can also consider an oral contraceptive pill in pre-menopausal women with amenorrhea and microprolactinoma.
 In a small study, no significant change in size of microadenomas was found after 2 years on an oral contraceptive pill.25
In a small study, no significant change in size of microadenomas was found after 2 years on an oral contraceptive pill.25
Growth Hormone–Producing Tumor (Acromegaly)
Growth hormone–producing tumor is a somatotroph cell adenoma producing classic physical features of acromegaly (gigantism in children). It is characterized by:
• Enlargement of facial features (74%) and hands and feet in adults (86%)
• Other features26,27 include cardiac disease (60%), excess perspiration (48%), arthralgia (46%), headache (40%), hypertension (39%), diabetes mellitus (38%), hypogonadism (38%), fatigue (26%), visual defect (26%), obstructive sleep apnea (13–50%), carpal tunnel syndrome (9%), colonic polyps (1.2% colonic cancer), multinodular goiter, benign prostatic hypertrophy, osteopenia, renal stones
 There is early cardiovascular mortality if growth hormone (GH) excess is untreated (34% die due to cardiovascular disease).
There is early cardiovascular mortality if growth hormone (GH) excess is untreated (34% die due to cardiovascular disease).
 Over 70% of tumors are macroadenomas at presentation.27
Over 70% of tumors are macroadenomas at presentation.27
Epidemiology
The incidence is 3 cases/million persons/year; the prevalence is 60/million.28
Pathophysiology
Benign monoclonal: multiple genetic mutations have been associated with GH-producing adenomas, including Gs-α stimulating mutations, growth arrest and DNA damage-inducible (GADD) loss of function mutations, PTTG protein.27
Differential Diagnosis
• Rarely due to somatotroph hyperplasia, GH, or GH-releasing hormone (GHRH) production from an ectopic tumor. More primitive cell origin (mammosomatotroph) may co-secrete prolactin.
• Can have plurihormonal adenoma and GH-cell carcinoma.
• Genetic syndromes:
 MEN1 (see above)
MEN1 (see above)
 Familial isolated pituitary adenomas (FIPA): 30 to 50% have AIP aryl hydrocarbon–interacting protein gene (AIP) mutation29
Familial isolated pituitary adenomas (FIPA): 30 to 50% have AIP aryl hydrocarbon–interacting protein gene (AIP) mutation29
 McCune-Albright syndrome: somatotroph hyperplasia due to Gs-alpha mutation in GHRH receptor; patients also have café-au-lait lesions, osteodystrophy, and other endocrine hyperfunctioning states (e.g., premature menarche, hyperthyroidism, adrenal Cushing’s)
McCune-Albright syndrome: somatotroph hyperplasia due to Gs-alpha mutation in GHRH receptor; patients also have café-au-lait lesions, osteodystrophy, and other endocrine hyperfunctioning states (e.g., premature menarche, hyperthyroidism, adrenal Cushing’s)
 Carney’s syndrome (see above)
Carney’s syndrome (see above)Table 9.2 Common conditions resulting in False-Positive and False Negative Testing in the Diagnosis of Acromegaly31
Conditions increasing IGF-1 levels |
Pregnancy* |
Thyrotoxicosis* |
Glucocorticoids |
Conditions decreasing IGF-1 levels |
Estrogen therapy* |
Hypothyroidism |
Fasting* |
Obesity |
Poorly controlled diabetes* |
*Also causes increased and insuppressible GH levels.
Diagnosis
• Screening test: elevated insulin-like growth factor-1 (IGF-1).
• Confirm with 2-hour 75-g oral glucose suppression test; GH remains > 1 µg/L.30
• Consider causes of false positives and false negatives if lab testing does not correlate with clinical findings31 (Table 9.2).
Medical Treatment
• Consider medical therapy with a somatostatin analogue:
 If patient is a poor surgical candidate due to high surgical risk from coexisting medical disorders.
If patient is a poor surgical candidate due to high surgical risk from coexisting medical disorders.
 If there is an invasive macroadenoma with no mass effect and low likelihood of surgical cure.30
If there is an invasive macroadenoma with no mass effect and low likelihood of surgical cure.30
 If patient is not cured by surgery, patients are more likely to respond if the tumor is densely granulated in the histopathological examination.32
If patient is not cured by surgery, patients are more likely to respond if the tumor is densely granulated in the histopathological examination.32
 Preoperative treatment with a somatostatin analogue in macroadenomas may increase surgical remission rates33–37; however, further studies are needed, so pretreatment is not routinely recommended unless surgery is delayed.
Preoperative treatment with a somatostatin analogue in macroadenomas may increase surgical remission rates33–37; however, further studies are needed, so pretreatment is not routinely recommended unless surgery is delayed.
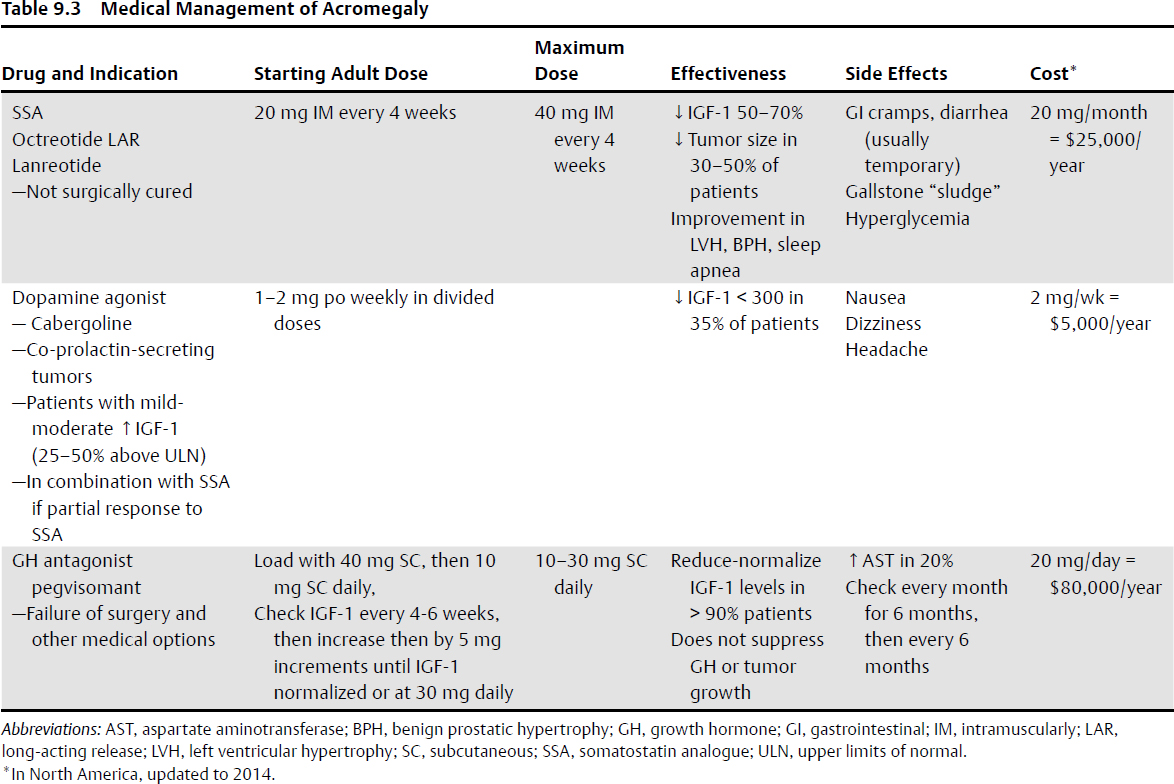
• Other medical treatment options include dopamine agonists and growth hormone receptor antagonists (see Table 9.3 for details about medical therapy for acromegaly).
• All patients should be screened for coexisting complications from acromegaly (with 2D echocardiogram, colonoscopy, hemoglobin A1C, fasting glucose and lipids, prostate-specific antigen [PSA], bone mineral density [BMD], consider thyroid ultrasound, consider sleep study, plasma calcium level).
Adrenocorticotropic Hormone (ACTH)-Producing Tumor (Cushing’s disease)
Cushing’s disease is the result of a corticotroph adenoma overproducing ACTH. The classic features include the following38:
• Central weight gain (95%), supraclavicular and dorsal fat pads, moon facies (90%), thin skin (85%), menstrual irregularities (80%), hypertension (75%), psychosis and depression (75%), bruising (65%), proximal muscle weakness (60%), glucose intolerance (60%), osteopenia or fractures (50%), violaceous striae, acne, hirsutism, male-pattern alopecia, hypokalemia, increased thromboembolic events
• Early mortality if cortisol excess remains untreated
Epidemiology
The incidence of Cushing’s disease is 1.2 to 1.7 cases/million/year.39
Pathophysiology
Pathophysiological findings include monoclonal expansion of corticotroph cells, usually microadenoma (80–85%), rarely hyperplasia, rarely familial (MEN1 is more typically a GH- or prolactin-producing tumor). Most tumors are densely granulated, basophilic, and often arise in the central part of the pituitary, due to the abundance of corticotrophs in that location.
Differential Diagnosis
• Exogenous steroids must be ruled out first.
• ACTH-dependent: pituitary adenoma or ectopic ACTH-producing tumor.
• ACTH-independent: adrenal adenoma, carcinoma, or hyperplasia.
• Pseudo-Cushing’s: caused by depression, severe stress, chronic alcoholism, or central obesity.
Diagnosis in Three Steps40
1. Demonstrate abnormal excess from the hypothalamic-pituitary-adrenal (HPA) axis: at least two of the following should be abnormal:
• Lack of suppression of HPA axis by low-dose dexamethasone suppression test (DST)
 1 mg overnight DST: dexamethasone is taken between 11 PM and midnight, or
1 mg overnight DST: dexamethasone is taken between 11 PM and midnight, or
 2 day low-dose DST (0.5 mg every 6 hours for 48 hours)
2 day low-dose DST (0.5 mg every 6 hours for 48 hours)
Either test is abnormal if the 8 AM cortisol is > 50 nmol/L after the last dose of dexamethasone.
• Overproduction of cortisol: elevated 24-hour urine for free cortisol (three- to fourfold elevation is very specific).
• Lack of diurnal variation of cortisol: elevated midnight salivary levels (cut point is assay-dependent).
Dexamethasone–corticotropin-releasing hormone (CRH) stimulation test can be done if the above results are inconclusive, especially if pseudo-Cushing’s is suspected.
2. Measure ACTH to check for ACTH dependence (should be > 4 pmol/L in ACTH-dependent Cushing’s).
3. With unsuppressed ACTH, decide if ACTH source is pituitary or ectopic (85–95% will be pituitary):
• MRI sella: tumor size > 6 mm is suggestive of pituitary etiology41,42 (also image the thorax/abdomen if ectopic source is suspected).
• Inferior petrosal sinus sampling (IPSS) to confirm pituitary source of ACTH (stimulated by CRH or deamino-8-D-arginine vasopressin [DDAVP] 10 µg IV), sample blood from the left and right sinuses and central vein for plasma ACTH at baseline and 0, 2, 5, 10, and 15 minutes after CRH or DDAVP given.
 In Cushing’s disease, expect central/peripheral gradient ratio > 2:1 (with no CRH or DDAVP) and > 3:1 (stimulated with CRH or DDAVP).42
In Cushing’s disease, expect central/peripheral gradient ratio > 2:1 (with no CRH or DDAVP) and > 3:1 (stimulated with CRH or DDAVP).42
 Lateralization with > 1.5:1 gradient between petrosal sinuses.
Lateralization with > 1.5:1 gradient between petrosal sinuses.
 Conflicting studies:
Conflicting studies:
 More accurate lateralization with IPSS than with MRI (70% vs 49%, p < 0.06).43
More accurate lateralization with IPSS than with MRI (70% vs 49%, p < 0.06).43
 When conventional MRI is negative or nondiagnostic, central venous sampling (CVS; cavernous and petrosal) is not as accurate in tumor localization as dynamic MRI (90% vs 52–65% with IPSS).44
When conventional MRI is negative or nondiagnostic, central venous sampling (CVS; cavernous and petrosal) is not as accurate in tumor localization as dynamic MRI (90% vs 52–65% with IPSS).44
The following testing is generally less helpful:
• High-dose DST (8 mg dexamethasone at midnight, followed by 8 AM plasma cortisol level) is less helpful because:
 There is considerable overlap between the two populations (pituitary vs ectopic).45
There is considerable overlap between the two populations (pituitary vs ectopic).45
 There are different published cutoffs for the 8 AM plasma cortisol for the diagnosis of pituitary source (< 140 nmol/L, versus > 50% suppression, versus > 70% suppression from baseline 8 AM cortisol).
There are different published cutoffs for the 8 AM plasma cortisol for the diagnosis of pituitary source (< 140 nmol/L, versus > 50% suppression, versus > 70% suppression from baseline 8 AM cortisol).
• Corticotropin-releasing hormone stimulation of ACTH and cortisol (ACTH should increase by > 50% 45 minutes after CRH in Cushing’s disease).46
• Caveats for testing:
 Pseudo-Cushing’s: depression/alcoholism/severe stress/severe obesity.
Pseudo-Cushing’s: depression/alcoholism/severe stress/severe obesity.
 False elevation in cortisol: increased cortisol-binding globulin (CBG) due to exogenous estrogen, pregnancy, hepatitis.
False elevation in cortisol: increased cortisol-binding globulin (CBG) due to exogenous estrogen, pregnancy, hepatitis.
 False-positive DST: drugs that alter dexamethasone metabolism such as phenobarbital, carbamazepine, and others.40
False-positive DST: drugs that alter dexamethasone metabolism such as phenobarbital, carbamazepine, and others.40
 Incidental pituitary or adrenal masses can be “decoys” and not represent the actual ACTH-producing tumor.
Incidental pituitary or adrenal masses can be “decoys” and not represent the actual ACTH-producing tumor.
 Adrenal hyperplasia can be ACTH dependent or independent.
Adrenal hyperplasia can be ACTH dependent or independent.
Medical Treatment
• Consider medical therapy if patient is a poor surgical candidate.
• Ketoconazole: A small retrospective study suggests that adequate presurgical treatment with ketoconazole or metyrapone in Cushing’s disease may be associated with suppressed postoperative cortisol concentrations and an increased long-term remission rate47; however, further prospective studies are needed.
• Pasireotide somatostatin analogue (SSA) inhibits ACTH secretion via somatostatin type 5 receptor subset48; it normalizes urine free cortisol in 25% of patients. Side effect: hyperglycemia.
It is critical to rule out hyperfunction due to overproduction of prolactin, growth hormone, or ACTH before surgery, as it can influence the choice of therapy, the preparation for surgery, and the need for hormonal coverage after surgery.
Nonfunctional Sellar Masses
Nonfunctional pituitary masses (adenomas and other pathologies) can present incidentally on imaging, with mass effect, hypopituitarism, or hyperprolactinemia due to stalk compression.
Pituitary Dysfunction in Extra-Adenoma Pathologies
These pathologies include cysts (e.g., Rathke’s cleft cyst), craniopharyngiomas, parasellar tumors, and sellar metastases.
• Hypothalamic masses such as craniopharyngiomas are more likely to present with diabetes insipidus (DI), along with other tertiary hormone defects.
• Presentations with CNs III, IV, V1, V2, and VI palsies are more likely secondary to pituitary apoplexy or other non-adenoma pathology (e.g., meningioma, lymphoma, or metastases).
Hormonal hypofunction must be identified and treated preoperatively for all sellar and parasellar lesions (secretory or nonsecretory), especially hypocortisolism, hypothyroidism, and antidiuretic hormone (ADH) deficiency.
Aggressive Pituitary Adenomas
Aggressive pituitary adenomas are defined from a clinical perspective. They have an aggressive clinical behavior, recur early and frequently, and resist conventional treatment, including radiotherapy.49
• Approximately 25 to 55% of pituitary adenomas are invasive,50–52 showing invasion of the surrounding structures, but the real percentage of aggressive pituitary adenomas is unknown.
• Suprasellar extension is not a criterion of invasiveness. Invasion refers to the bone erosion-invasion (Hardy grades III and IV; see Chapter 21, Fig. 21.4, on page 500), cavernous sinus invasion (Knosp grades 3 and 4; see Fig. 21.5, on page 501), or invasion of the sphenoid sinus (also shown in histopathology by the invasion of the sinus mucosa).
• Aggressive pituitary adenomas are nonmetastatic, but can share common histological features with pituitary carcinomas.49
• Typically they show atypical histological features, such as high mitotic activity, Ki-67 ≥ 3%, or extensive p53 immunopositivity.53
• Some histotypes of pituitary adenomas have been associated with a higher incidence of clinical aggressiveness. Some pituitary adenoma subtypes associated with clinical aggressiveness include:
 Corticotroph tumors containing Crooke cells (cells that undergo massive accumulation of perinuclear cytokeratin in the presence of excess glucocorticoid); 60% of patients with Crooke cell tumors had recurrences at > 1 year follow-up (multiple recurrences in 24%). Average time to recurrence: 3.6 years.54
Corticotroph tumors containing Crooke cells (cells that undergo massive accumulation of perinuclear cytokeratin in the presence of excess glucocorticoid); 60% of patients with Crooke cell tumors had recurrences at > 1 year follow-up (multiple recurrences in 24%). Average time to recurrence: 3.6 years.54
 Silent corticotroph adenomas: tumors morphologically similar to ACTH-adenomas, but with no clinical or biochemical evidence of hormone excess.55 Silent corticotroph adenomas, especially the subtype 3,56 have been associated with clinical aggressiveness, although this correlation is controversial.57
Silent corticotroph adenomas: tumors morphologically similar to ACTH-adenomas, but with no clinical or biochemical evidence of hormone excess.55 Silent corticotroph adenomas, especially the subtype 3,56 have been associated with clinical aggressiveness, although this correlation is controversial.57
 Acidophil stem cell adenomas: rapidly growing plurihormonal adenoma, undifferentiated with focal immunopositivity to GH and prolactin (PRL).
Acidophil stem cell adenomas: rapidly growing plurihormonal adenoma, undifferentiated with focal immunopositivity to GH and prolactin (PRL).• Patients affected by aggressive pituitary adenomas should undergo strict biochemical and radiological follow-up and multimodal treatment, including surgery, radiotherapy/radiosurgery, and/or chemotherapy (temozolomide).49
Nelson’s Syndrome
• Aggressive corticotroph tumor growth after bilateral adrenalectomy for Cushing’s disease presumably due to lack of feedback inhibition after adrenals are resected.
• Occurs after bilateral adrenalectomy in approximately 8 to 43% of patients58; 21% of patients in a recent systematic review.59 There is a higher incidence among children.
• A small study suggests that stereotactic radiosurgery of pituitary tumors before bilateral adrenalectomy may decrease the incidence of Nelson’s syndrome.60
Diagnosis
• Hyperpigmentation, mass effect from tumor.
• Check ACTH at 8 AM prior to steroid dose:
 High ACTH levels (> 110 pmol/L) in addition to progressive elevation of ACTH on three occasions is diagnostic.58
High ACTH levels (> 110 pmol/L) in addition to progressive elevation of ACTH on three occasions is diagnostic.58
• MRI of the sella (3 months postadrenalectomy, then every 6 months for 2 years, then yearly or if clinically indicated).58
Management of Nelson’s Syndrome
• Surgery is first-line.
• Adjuvant radiotherapy: consider in patients with remnant tumor after transsphenoidal surgery.
• Stereotactic radiotherapy: data are limited and conflicting, with one study showing remission in 14%,61 whereas another study showed no tumor re-growth at 7 years post–stereotactic radiosurgery.62
• Medical therapy (limited options):
 Dopamine agonists: cabergoline can induce remission of Nelson’s syndrome and tumor shrinkage.63–65
Dopamine agonists: cabergoline can induce remission of Nelson’s syndrome and tumor shrinkage.63–65
 Selective somatostatin analogue: case report using pasireotide long-acting release (LAR) resulted in a significant decrease in ACTH level and reduction in tumor size.66
Selective somatostatin analogue: case report using pasireotide long-acting release (LAR) resulted in a significant decrease in ACTH level and reduction in tumor size.66
 Temozolomide: alkylating agent may be effective, but more studies are needed.67
Temozolomide: alkylating agent may be effective, but more studies are needed.67
Pituitary Carcinoma
Epidemiology
Pituitary carcinoma accounts for 0.1% to 0.2% of pituitary tumors, defined as a pituitary tumor with craniospinal or systemic metastases.68
• Poor prognosis: 66% mortality in the first year of diagnosis.69
• Many pathological features overlap with atypical pituitary tumors.
• Prolactin-secreting pituitary carcinoma, corticotroph pituitary carcinomas, and growth hormone secreting carcinomas can occur.70
• There have been few reports of gonadotroph carcinomas, and thyroid-stimulating hormone (TSH)-secreting pituitary carcinomas are the most uncommon subtype.71–73
• Often, the clinical presentation is similar to that of adenomas, but pituitary carcinoma may be unresponsive to standard therapies and may recur quickly.
• 38.7% have systemic metastases, 45.2% craniospinal metastases, 16.1% have both.74
• Metastatic pituitary carcinoma from breast/lung primary accounts for 1% of all pituitary cancers.
Pathophysiology
Molecular mechanisms for progression from adenoma to carcinoma remain unclear.
• Molecular markers for an aggressive tumor: current markers have limitations
 Ki-67 > 3% used to denote atypical pituitary tumor by World Health Organization (WHO) criteria,53 but not all studies have demonstrated a difference in Ki-67 between pituitary carcinomas and other categories of adenomas.
Ki-67 > 3% used to denote atypical pituitary tumor by World Health Organization (WHO) criteria,53 but not all studies have demonstrated a difference in Ki-67 between pituitary carcinomas and other categories of adenomas.
 p53 immunoreactivity correlates with pituitary tumor invasiveness.75
p53 immunoreactivity correlates with pituitary tumor invasiveness.75
Treatment
• Surgery
• Radiation to prevent regrowth in partially resected tumors
• Medical therapy:
 Control biochemical secretion: similar treatment to pituitary adenomas
Control biochemical secretion: similar treatment to pituitary adenomas
 Chemotherapy: data limited as there are no randomized studies
Chemotherapy: data limited as there are no randomized studies
 Most commonly reported agents used: cyclo-hexyl-chloroethyl-nitrosourea (CCNU; lomustine) and 5-fluorouracil until recently70
Most commonly reported agents used: cyclo-hexyl-chloroethyl-nitrosourea (CCNU; lomustine) and 5-fluorouracil until recently70
 Current first-line therapy: temozolomide, with favorable response in 11 of 16 (69%) patients76
Current first-line therapy: temozolomide, with favorable response in 11 of 16 (69%) patients76
 Special Situations
Special Situations
Pregnancy
In pregnancy, a normal pituitary increases by 30% in volume and 2.6 mm in height, due to hyperplasia of lactotrophs.77
• For intrasellar prolactinomas, usually stop dopamine agonist at conception.
• Risk of growth of prolactinoma in pregnant women:
 Microadenoma: 0 to 1.3%
Microadenoma: 0 to 1.3%
 Macroadenoma: 10 to 25%; after surgery 3%78
Macroadenoma: 10 to 25%; after surgery 3%78
• Macroprolactinomas that extend into the suprasellar region are treated with bromocriptine (or cabergoline) throughout pregnancy.78
• There are challenges to diagnose pituitary hyperfunction during pregnancy (prolactin, GH, or ACTH overproduction) due to normal physiological changes.
• New diagnoses of Cushing’s are usually adrenal in origin in pregnancy.79
• It is challenging to assess for adrenal insufficiency (the morning cortisol in the third trimester rises to double the level—420 vs 980 nmol/L—in healthy women).
• For patients with hypofunction of the pituitary, the dose of L-thyroxine is typically increased by 30% in the first trimester, and the dose of hydrocortisone may need to increase slightly in the third trimester, to adjust for the normal physiological changes in each axis during pregnancy.
• One can try a dopamine agonist to reduce the size of normal lactotrophs and to reduce chiasmal compression when the combined mass of hyperplastic pituitary and tumor compromises the optic chiasm.
• Placental vasopressinase in third trimester can precipitate borderline diabetes insipidus.
Indications for Surgery
Surgical Indications for Mass Effect
• Visual compromise (decreased acuity or visual field defects), diplopia, headaches, or facial numbness in V1 or V2 distribution are indications for surgery.
• Prolactinomas usually respond to medical therapy with dopamine agonists even when there is significant mass effect.80–82
• Management of pituitary apoplexy is urgent surgery when there is visual compromise. Diplopia and headache may resolve spontaneously without surgical intervention.83 See also Chapter 35, page 859.
Surgical Indications for Hyperfunction
Prolactinomas
• Medical treatment is first line (as above).
• Surgery is appropriate in symptomatic patients who cannot tolerate high doses of dopamine agonists or in those who do not respond to dopamine agonists.8
Acromegaly
• Surgery is the first-line therapy for all patients with surgically curable adenomas and for those with macroadenomas giving rise to mass effect.30
• Can consider surgery in those with macroadenomas without mass effect and a low likelihood of surgical cure to improve response to medical treatment.30
Cushing’s Disease
• Surgery is first-line therapy for most patients, even in macroadenomas or invasive adenomas.84
Surgery and Pituitary Hypofunction
• Hypofunction may improve after surgery.
 Improvement may be due to the restoration of normal hypothalamic-pituitary portal circulation and decrease in pituitary stalk compression.85
Improvement may be due to the restoration of normal hypothalamic-pituitary portal circulation and decrease in pituitary stalk compression.85
 If there has been ischemic necrosis or destruction of the gland by tumor, then improvement in hormonal function is unlikely.
If there has been ischemic necrosis or destruction of the gland by tumor, then improvement in hormonal function is unlikely.
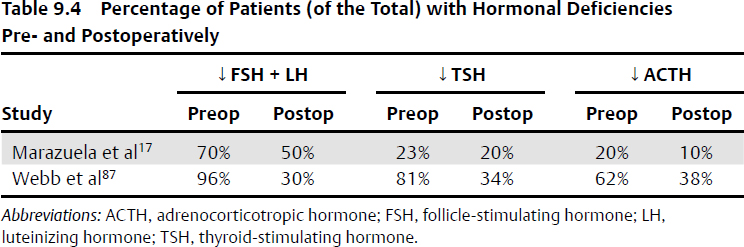
• 46 to 65% of patients had recovery of between one to three hormonal axes after surgery17,86,87 (Table 9.4).
• In one study of 234 patients,87 40% had preoperative hormonal deficit. Postoperatively, 22% had a new hormonal deficit (3–4% in other studies), and of those with preoperative hormonal deficit, 45 (48%) recovered from one to three deficiencies.
• A more recent meta-analysis found fewer than one third of patients with hormonal deficiencies recovered after transsphenoidal surgery.88
Preoperative Considerations
Baseline Tests for Hyper/Hypofunction
• Prolactin: need serial dilutions to rule out hook effect if it is a macroadenoma.