Figure 1.1. Prevalence bias. Each horizontal line represents a case with active disease (i.e., a prevalent case). The length of the line represents the time of the active disease, with onset to the left and offset or death to the right. The dashed vertical line represents the day on which prevalence is measured. Long-duration cases are oversampled (8 of 8 are ascertained on the prevalence day) relative to short-duration cases (2 of 7 are ascertained on the prevalence day).
Epidemiologic investigations since these early studies continue to inform and challenge our understanding of epilepsy. This chapter aims to outline current definitions and distinctions in epidemiologic research. In addition, findings from more recent studies and the challenges presented to investigators conducting these studies are outlined.
FREQUENCY MEASURES OF INCIDENCE AND PREVALENCE
Incidence is expressed as the number of new cases of disease in a standard-sized population per unit of time—for example, the number of cases per 100,000 population per year. Prospective studies of incidence are advocated as they permit observation of any changes in the incidence rate (20) and may therefore identify risk factors that play a causal role in the development of epilepsy (21,22). Ongoing surveillance studies of this type, however, are time consuming and costly (20,23) and are therefore less common than prevalence studies (20,23) and less likely to be conducted in resource-poor countries (24). Incidence rates for epilepsy are typically between 30 and 80 per 100,000 population per year in developed countries but have been observed to exceed these figures. Table 1.1 presents a selection of studies illustrating this trend.
Table 1.1 Incidence and Prevalence of Epilepsy as Reported in Selected Population-Based Studies Throughout the World
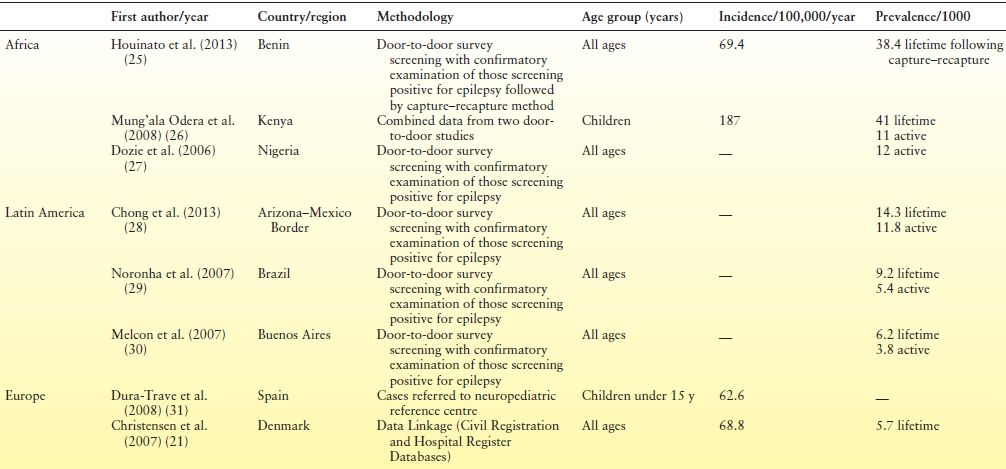
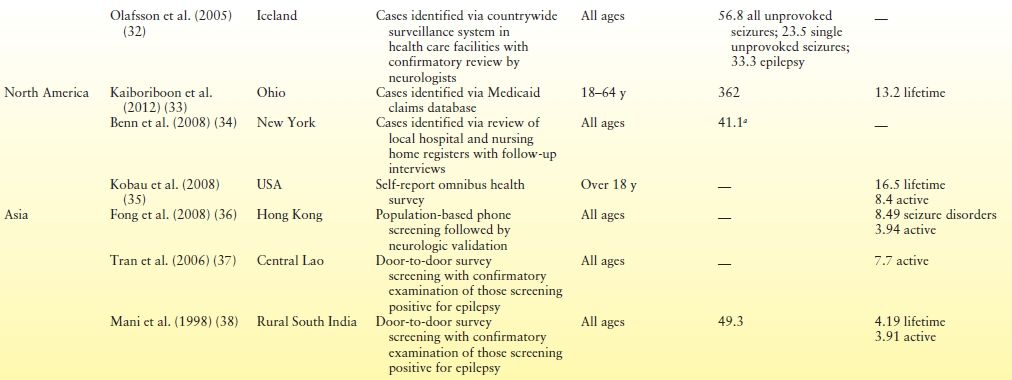
aFirst unprovoked seizure and newly diagnosed epilepsy.
Worldwide, rates vary across regions, with rates being typically higher in resource-poor countries (22), most especially in Africa and Latin America, where figures can exceed 100 per 100,000 population (39–41). Rates also differ by age, but differentially in developed and resource-poor countries. In developed countries, rates are highest among infants and older persons (31,42–44). Although incidence rates among children have fallen over the last three decades in developed countries, this decrease has been offset by an increase among older persons (45). A different age-related pattern emerges in developing countries, however, where a decrease in incidence is observed with age (22). The larger proportion of children in the population in developing countries is thought is to contribute to the higher overall incidence rates when compared with developed countries (21,22).
Prevalence studies measure the total number of persons with epilepsy at a specific moment in time. Prevalence rates are usually expressed as the number of persons with epilepsy per 1000 population. Estimates of active epilepsy are typically the focus of prevalence studies, with those in remission or who are not receiving treatment at the time of case ascertainment being excluded. A “plethora of studies” that consistently report prevalence estimates of active epilepsy in developed countries of between 4 and 10 per 1000 population suggest that there is “little justification for further cross-sectional studies of prevalence” (46, p. 168) in these countries. Recent findings from Norway, however, of 12 per 1000 treated epilepsy and 7 per 1000 active cases in a population that excluded high-risk groups such as older persons have led investigators to suggest that the true prevalence of epilepsy in developed countries may be higher than previously reported (47). Prevalence estimates typically increase with age and are generally higher among males than females (20) although this difference may not always reach statistical significance.
Prevalence estimates in resource-poor countries are generally higher than in developed countries (46). Median prevalence of active epilepsy in Latin America is reported as 12.4 per 1000 population; however, this finding conceals widely varying findings from individual studies ranging 5.1 to 57 per 1000 population (48). This wide variation in estimates is also observed in Africa where estimates have been reported ranging from 5.2 to 58 per 1000 (49,50). While researchers note that known risk factors, such as environment, contribute to the high prevalence estimates on the African continent (26), some authors suggest that the true prevalence estimate may actually be higher again as disclosure of the condition is particularly problematic (51). Reviews of studies conducted in Asia, perhaps surprisingly, report findings aligning more closely with those in developed countries. This has led some investigators to speculate whether there is a specific protective factor as yet unknown in Asia or whether the finding reflects specific risk factors in Latin America and Africa (23). Table 1.1 presents findings from some recent studies worldwide.
Several recent studies have examined the relative frequency, if not absolute incidence, of different forms of epilepsy in well-characterized series of incident patients who were reasonably representative of the populations from which they were drawn (Table 1.2). Apart from the obvious difference between adults and children, there is a degree of variation among the studies just of children as well. Whether this represents real differences across populations or methodologic difference between studies is not clear. Certainly, patterns of referral to recruitment sources as well as diagnostic ability of the physicians who evaluate the patients could create apparent difference between studies where none exist. Such concerns aside, a few generalities can be drawn. Fewer children than adults are likely to have an unclassified form of epilepsy. In children, idiopathic focal epilepsies (largely dominated by benign rolandic epilepsy) comprise about 5% to 10% of childhood-onset epilepsy. The idiopathic generalized epilepsies comprise 20% to 40%. Finally, between 10% and 20% of childhood-onset epilepsy falls into the category of secondary generalized. These are some of the most devastating and intractable forms of epilepsy and include West and Lennox–Gastaut syndrome.
Table 1.2 Distribution of Epilepsy Syndromes in Newly Diagnosed Patients from Eight Different Countries
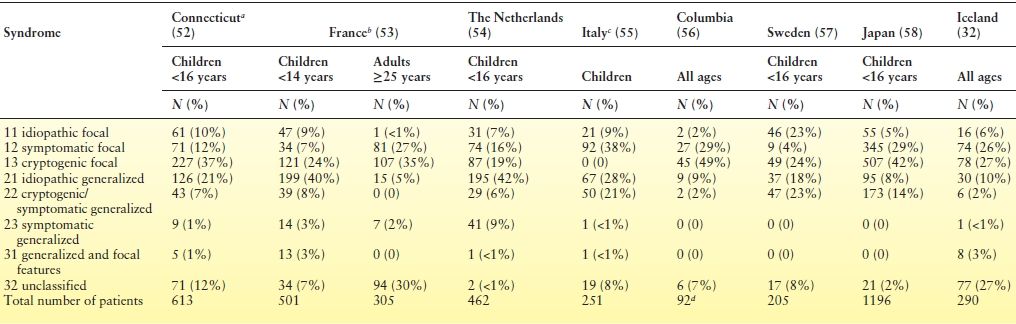
aThe cryptogenic and symptomatic localization–related categories were redefined to be consistent with the interpretation of other authors and to facilitate comparisons.
bLimited to children <14 y of age.
cPediatric epilepsy center (referral) in Milan—published prior to 1989.
dListed as “special syndrome.”
CURRENT DEFINITIONS AND DISTINCTIONS USED IN EPIDEMIOLOGIC EPILEPSY RESEARCH
Epilepsy (recurrent, unprovoked seizures) must be distinguished from many other conditions and situations in which seizures may occur. The following definitions are generally accepted and are in widespread use.
Epileptic Seizure
An epileptic seizure is a “transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain” (59). Epileptic seizures must be distinguished from nonepileptic seizures and from other conditions that may produce clinical manifestations that are highly similar to those caused by epileptic seizures.
Acute Provoked Seizure
An acute provoked seizure is one that occurs in the context of an acute brain insult or systemic disorder, such as, but not limited to, stroke, head trauma, a toxic or metabolic insult, or an intracranial infection (60).
Unprovoked Seizure
A seizure that occurs in the absence of an acute provoking event is considered unprovoked (60). A history in the past of stroke, trauma, or other condition may be present; however, at the time of the seizures, no specific acute insult has occurred.
Epilepsy
The widely accepted operational definition of epilepsy requires that an individual has at least two unprovoked seizures on separate days, generally 24 hours apart. An individual with a single unprovoked seizure or with two or more unprovoked seizures within a 24-hour period is typically not at that time considered to have met the criteria for the diagnosis of epilepsy per se (60). An International League Against Epilepsy (ILAE) document from 2005 attempted to provide a conceptual definition of epilepsy, which entailed the notion of an enduring underlying predisposition to unprovoked seizures (59). The definition was presented, however, as an operational definition and engendered considerable controversy and response (61–63) precisely because there was no way to ensure that it would be consistently and validly applied across different settings by different investigators, a quality that is a prerequisite for meaningful research. A new practical clinical definition of epilepsy has now been agreed upon. Epilepsy is defined as any one of the following conditions: (a) at least two unprovoked (or reflex) seizures occurring >24 hours apart; (b) one unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years; or (c) diagnosis of an epilepsy syndrome. Epilepsy is considered to be resolved for individuals who either had an age-dependent epilepsy syndrome but are now past the applicable age or have remained seizure free for the last 10 years and off antiseizure medicines for at least the last 5 years (63). As this definition was just published in 2014, few studies have used it, and most of the literature is based on the earlier definition of two or more unprovoked seizures.
A first seizure often is the first identifiable sign of epilepsy, and in some cases, it is possible to recognize the specific underlying disorder (form of epilepsy) at its earliest presentation (53). In the case of Dravet syndrome, the first definitive sign may be a febrile seizure—typically hemiclonic and prolonged—and, with a genetic test, the epilepsy may be diagnosed before “unprovoked” seizures emerge (64). Such situations may be rare and are difficult to ascertain and handle well in many epidemiologic studies because of the relatively crude diagnostic information used. For the field of epilepsy, however, they are increasingly important and should not be ignored.
Etiology
Terminology for referring to etiology has evolved considerably in the last several years. The old, traditional terms, idiopathic, cryptogenic, and symptomatic, are gradually being abandoned in favor of clearer more descriptive language. The 2010 ILAE Commission report recommended as an initial starting point the terms “genetic,” “structural–metabolic,” and “unknown” to refer to classes of causal factors. Note, these are categories for causes, not the form (syndrome) of epilepsy per se. Each was carefully defined: genetic refers to causes for which, to our best understanding, the seizures are a direct result of the genetic error and seizures are the core symptom of the disorder. Evidence concerning a genetic role may come from molecular studies but may also be derived from well-conducted clinical studies (e.g., twin studies). This differs from the old idiopathic (meaning “presumed genetic”) as a basis for the genetic inference is stated, and, unlike idiopathic, genetic includes disorders that do not necessarily have a “benign” outcome. The unwieldy term “structural–metabolic” is intended to identify people with known underlying brain disorders that are primary contributors to their epilepsy. As most metabolic and many structural disorders have a genetic basis, the distinction between genetic–structural–metabolic may seem fuzzy. The distinction focuses on the mechanism by which the genetic defect influences epilepsy as well as the constellation of symptoms presented by the patient, primarily seizures or seizures secondary to some other medical condition. The role of autoimmunity, an extremely important contributor to some epilepsies is not adequately evoked by the term structural–metabolic either (65,66). As the mechanisms linking the precipitating cause to development of epilepsy become better understood, it becomes more difficult to pigeonhole some causes. For example, recent evidence suggests that some forms of cortical malformations that cause epilepsy are themselves due to viral infections in utero (67). Finally, for referring to situations in which the cause is simply unknown, the term “unknown” was suggested. This was meant to be considered as a true statement of uncertainty or ignorance, without assumptions.
The ability to identify the likely causes of epilepsy has changed dramatically over time with the introduction and increasing sophistication of neuroimaging and genetic testing. These changes are not uniformly implemented across countries or even across subpopulations within a country. Consequently, the unevenness of precision raises problems for cross population (including across time period) comparisons. For epidemiologic purposes, especially on the global level, a very pragmatic distinction could be made between what might be called complicated and uncomplicated epilepsy. Complicated epilepsy would include any epilepsy associated with a factor that is presumed to cause epilepsy (stroke, trauma, tuberous sclerosis, etc.) or any evidence of neuroimpairment (substantial developmental delay, intellectual impairment, abnormal motor or sensory exam) that can result from a cortical insult. Implicitly, uncomplicated would refer to epilepsy in which there is no clear insult or condition to which the occurrence of the epilepsy can be attributed and the individual is neurotypical (normal exams and cognition).
Grey Areas
Neonatal seizures (i.e., those occurring in an infant <28 days old) are usually differentiated from epilepsy for a variety of reasons; however, this approach makes little sense as several specific forms of epilepsy have been reported in this age group (68), and there is no good reason that factors that would be considered structural or metabolic causes of epilepsy in an older individual would be treated differently in the neonate. Unfortunately, epidemiologists often do not have the detailed information needed to distinguish the child with a seizure secondary to transient hypoglycemia from a child who seizes because of a KCNQ2 encephalopathy (69) or a brain malformation. Febrile seizures are a well-described and recognized seizure disorder, which, for historical reasons, has been distinguished (both clinically and in research) from epilepsy. For those involved in detailed genetic investigations, this may be an inappropriate distinction. For the epidemiologist, however, who may not always have the necessary clinical and particularly the genetic detail, the distinction is of value. It also has important clinical implications for the treatment of most of these children with such seizures.
Epilepsy Syndromes
Epilepsy syndromes have been alluded to above and are presented in greater detail in subsequent chapters of this book. Epilepsy, like cancer, is not a single disorder, and the efforts to identify specific forms of epilepsy reflect the importance of the diversity within the epilepsies. The epilepsy syndromes represent forms of epilepsy that have different causes, different manifestations, different implications for short- and long-term management and treatment, anticipatory guidance, genetic counseling, and long-term outcomes. Many epidemiologic studies do not attempt to identify specific forms of epilepsy; however, in large-scale population- and community-based studies, it is possible to do so, provided the investigators have access to the necessary information and the expertise needed to diagnose these syndromes (52–54). As the questions in epileptology become increasingly sophisticated, including precise characterization of specific causes, types of seizures, and types of epilepsy, the gap between what typical epidemiologic studies can do versus the types of information needed from them has been widening.
Seizure Types
Perhaps the greatest and most relevant distinction for broad-scaled epidemiologic investigations is between convulsive and nonconvulsive seizures as a very crude marker of severity. The term tonic–clonic should generally be eschewed in epidemiologic studies as it is often misused to refer to any “big” seizure regardless of whether tonic and clonic components are both present. Diagnostic precision to identify other seizure types is generally inadequate in epidemiologic studies, and the basis of identifying other seizure types requires significant scrutiny.
EPIDEMIOLOGIC CHALLENGES
Epidemiologic studies have provided valuable insights into the frequency of seizures within the population and have provided the initial impetus for some of the distinctions outlined above. As Kurland noted, however, epidemiologists need to be vigilant to potential sources of bias that threaten the validity of their findings. The ability of diagnosticians to appropriately identify cases and the capabilities of epidemiologists to identify those cases within the population are fundamental issues within the field of epidemiology. We also note that, as the clinical and scientific field of epilepsy has grown much more sophisticated, epidemiologists must work to keep up with important diagnostic and lexical distinctions.
Diagnostic Issues and Considerations in Ascertaining Cases
Seizures and epilepsy present a complex situation because the diagnosis is not based on a single source or type of information. Rather, epilepsy is a clinical diagnosis supported to a greater or lesser extent by a wide range of data obtained from several sources: the history (both from the patient and witnesses) of the events believed to be seizures, the circumstances under which the events occur, the past medical history, medications, a neurologic examination, reliable EEG, and increasingly neuroimaging (70). To have a valid diagnosis, one must also be able to rule out many other conditions that mimic seizures. These disorders include, but are not limited to, movement disorders, parasomnias, attention deficit/hyperactivity disorder, pseudo- or nonepileptic seizures, transient ischemic attacks, and syncope (71,72).
Ideally, a diagnosis of epilepsy should be undertaken by medical practitioners with expertise in epilepsy (73). Unfortunately, access to neurologists and epilepsy specialists is generally poor in developing countries and often poor in developed countries as well. Consequently, diagnoses may be made by those with only minimal expertise in the field (35,74). Estimates of misdiagnosis rates suggest that over one-fifth of persons with a diagnosis of epilepsy may be misdiagnosed (75,76). Reevaluation of initial diagnosis of epilepsy in epidemiologic studies reports rates of 23% (77,78) with diagnostic doubt among patients diagnosed by neurologists and nonspecialists reported at 5.6% and 18.9%, respectively (79). In 2007, a UK All Party Parliamentary Group on Epilepsy convened to determine the “human and economic cost of epilepsy” in England. The report estimates that 74,000 people in England are misdiagnosed with epilepsy and are therefore receiving inappropriate treatment. The financial cost of unnecessary or incorrect treatment combined with lost employment was estimated in 2007 at £134 million per annum (80).
Epidemiologic studies that rely on medical registers for case ascertainment provide valuable insights into levels of misdiagnoses. Christensen et al. (21), for example, randomly selected 200 patients with an ICD diagnosis of epilepsy from the Danish National Hospital Register, a national register of all discharges and outpatient cases from Danish hospitals. The authors found that almost one in five (19%) of the sample did not fulfill the ILAE criteria for an epilepsy diagnosis. In fact, approximately 7% of patients were given an epilepsy diagnosis on the basis of one seizure. Christensen et al. (81) also noted that while the validity of epilepsy diagnosis from the register was moderate to high, there was low predictive value for epilepsy syndromes.
Primary care registers, a common source of case ascertainment in epidemiologic research, have also been found to include persons incorrectly diagnosed with epilepsy. Gallitto et al. (82) gathered population-based data from general practitioners (GPs) in the Aeolian Islands. All established or suspected cases of epilepsy were evaluated by epileptologists in the local outpatient services with the support of the local GPs or, for those with additional disabilities, within the family home. The evaluations comprised a review of medical notes and where necessary EEG or neuroradiologic investigation. Following the epileptologic evaluation, 30% of established and suspected cases were identified as not fulfilling the diagnostic criteria for epilepsy.
These high levels of misdiagnosis have resulted in calls for a gold standard diagnostic criterion to distinguish epilepsy from other conditions with similar clinical features (75). The UK National Institute for Health and Clinical Excellence (NICE) provides support to diagnosticians via its recently updated guidelines “The epilepsies; the diagnosis and management of the epilepsies in adults and children in primary and secondary care” (83). The NICE recommends that all individuals with a recent-onset suspected seizure be seen within a 2-week period by a specialist, defined as a medical practitioner with expertise in epilepsy. The implementation of these guidelines for children is currently being monitored by Epilepsy12, a nationwide audit aiming to ensure a standardized level of provision across the United Kingdom (84).
In addition to the determination of whether or not someone has epilepsy, adequate information is needed to identify the specific form of epilepsy and its underlying cause. While this level of detail is frequently absent from traditional epidemiologic studies, it must be incorporated in the future if epidemiologic studies are to continue to inform scientific and clinical endeavors relevant to epilepsy as it is understood and treated today. Without a meaningful diagnostic evaluation, epidemiologic studies can do little more than provide an approximate head count, which previous work has shown to be rather error prone. The lumping together of highly diverse disorders that share the diagnostic label “epilepsy” also limits the ability of epidemiologic studies to provide meaningful prognostic information.
The case ascertainment options described above, based on medical registers, may not be suitable for all epidemiologic studies. Where these registers are unavailable, or may be considered unrepresentative, other more population-based methods may be used to identify people with epilepsy. As was observed for medical registers, these methods also have their own unique challenges. Screening questionnaires, for example, are a common tool used in epidemiologic studies. Methodologies employing screening questionnaires typically comprise two phases. In the first phase, a screen is used to identify positive cases. In the second phase, these positive cases are evaluated clinically to confirm the presence of epilepsy. Noronha et al. (29), for example, used a screening tool developed by Borges et al. (85) in the first phase of their study to determine the prevalence of epilepsy in Brazil. The screening tool reported sensitivity and specificity at 96% and 98%, respectively. Similarly, Melcon et al. (30) used a modified version of a screening tool from the Copiah County study (86) to identify potential cases for inclusion in their prevalence estimate of epilepsy in Argentina. This screening tool also reported acceptable levels of sensitivity and specificity at 95% and 80%, respectively. More recently, a three-stage survey methodology has been proposed that comprised a two-item first survey, a more extensive second survey, and a third-stage clinical validation. This methodology reported a sensitivity of 49% and specificity of 100% and was found to be 37% less expensive than the more traditional two-stage process (87).
Screening tools are advocated by the World Health Organization, whose “Global Campaign Against Epilepsy” supports those undertaking epidemiologic research in resource-poor countries. Demonstration projects managed under this program, in addition to assessments of local knowledge, attitude, and health service provision, undertake epidemiologic door-to-door studies to determine prevalence estimates. Screening tools developed by WHO (88) have been used in large-scale national epidemiologic studies (56). Notwithstanding the successful application of screening tools in many studies, the diagnostic sensitivity and specificity of these tools can, however, be poor, and the training of physicians or other health care professionals charged with validating positive screens may be compromised by poor access to such basic diagnostic tools as EEGs.
Other case ascertainment sources used in epidemiologic studies include prescription databases recording antiepilepsy drug usage. By definition, these epidemiologic studies estimate “treated epilepsy” and are more common in developed countries where the treatment gap is minimal. Prescription databases have been found to offer a suitable means by which the prevalence of epilepsy can be determined in community samples (89) as the coverage of the databases is typically far broader than medical registers. Purcell et al. (90), for example, examined rates of treated epilepsy in the United Kingdom using the General Practitioner Research Database, which provided data on prescription use of over 1.4 million persons. More recently, D’Souza et al. (91) estimated rates of treated epilepsy in Tasmania via a two-stage process of firstly, identifying all residents in Tasmania who were supplied with at least one antiepilepsy drug prescription recorded on the national prescription database over a given time period and secondly, by following up on these individuals by a postal survey to determine whether or not they were receiving this treatment for epilepsy.
A potential source of bias in identifying persons with epilepsy from prescription databases is that cases cannot be clinically validated (90,92). This bias is magnified in situations where diagnosis is not recorded on the database and where “estimates” of drug use among people with epilepsy are applied (93,94). While antiepilepsy drugs have been previously identified as “tracers” of epilepsy due to their chronic and highly specific usage (94
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


