Epilepsies with Predominantly Myoclonic Seizures
Myoclonic seizures are a common feature of many types of epilepsy at all ages, and they are, therefore, a nonspecific manifestation. Epilepsies characterized by “true” myoclonic seizures can occur at any age. Those syndromes in which myoclonic attacks are the exclusive or predominant type of seizure are considered in this chapter, and they differ in outlook and therapy from the Lennox-Gastaut syndrome (LGS). Therefore, properly classifying the different types of seizures is important; this involves not only careful questioning of parents and caregivers but also the use of polygraphy and/or video-electroencephalograms (video-EEGs) when they are available. Video and neurophysiologic monitoring often reveals that different seizure types that are often referred to as myoclonic in the clinical histories because they involved a brisk movement are in fact brief tonic or atonic attacks. The precise determination of the other seizure types that often accompany myoclonic attacks is also important because the associated seizures are critical for classifying the myoclonic epilepsies.
In this chapter, after discussing the nosology and pathophysiology of myoclonic seizures, the authors review the nosologic and semantic problem of the myoclonic epilepsies and define the features common to all forms of myoclonic epilepsy, as well as the features of the different members of this heterogeneous group.
GENERAL NOTIONS ON THE MYOCLONIC EPILEPSIES
Definitions
The term myoclonus encompasses a group of neurophysiologically diverse phenomena of heterogeneous etiology whose common semiologic characteristic is represented by involuntary jerky movements that most commonly involve antagonist muscles (Patel and Jankovic, 1988). Myoclonus can be classified as “epileptic” and “nonepileptic” according to its physiology. Some authors define epileptic myoclonus as that occurring within the setting of epilepsy (Patel and Jankovic, 1988), while others define the term as those forms in which the underlying neurophysiologic substrate is thought to be a paroxysmal depolarization shift, regardless of which population of neurons (cortical or subcortical) is primarily involved (Hallett, 1985). Guerrini et al. (2002b) suggested that epileptic myoclonus can be comprehensively defined as an elementary electroclinical manifestation of epilepsy involving descending neurons whose spatial (spread) or temporal (self-sustained repetition) amplification can trigger overt epileptic activity. According to its distribution, myoclonus can be classified as focal, multifocal, or generalized (Hallett, 1985).
The electroencephalographic (EEG) correlate of generalized epileptic myoclonus is a generalized spike-wave discharge, in which the spike corresponds to the myoclonic jerk (Fig. 6.1) and the following slow wave to a postmyoclonic muscular silent period. The EEG correlate of focal epileptic myoclonus is a focal spike in the contralateral sensorimotor cortex. Sometimes, such a correlate can be detected only by the use of jerk-locked (EEG or magnetoencephalogram) averaging. The duration of the “positive” myoclonic muscular burst ranges between 10 and 100 milliseconds. Antagonist muscles are involved simultaneously. The postmyoclonic silent period (200 to 300 milliseconds) is usually proportional to that of the myoclonic jerk. Epileptic myoclonus can either be spontaneous, or it can be evoked by sensorial stimulation such as tapping, muscle stretching, or photic stimulation.
Epileptic myoclonus can also be classified as “negative” (Guerrini et al., 1993b) when the jerking is mainly related to brief pauses of ongoing electromyographic (EMG) activity that lasts 50 to 400 ms. These silent periods are time locked to a contralateral spike-wave complex that is usually central or frontocentral (Tassinari et al., 1995; Guerrini et al., 1993b). Negative myoclonus is characterized by a brief involuntary lapse of posture, followed by a jerk that results from the individual’s voluntary attempt to resume his or her original position (Fig. 6.2). Positive and negative myoclonus can be focal or generalized. However, generalized purely “negative” epileptic myoclonus is rare.
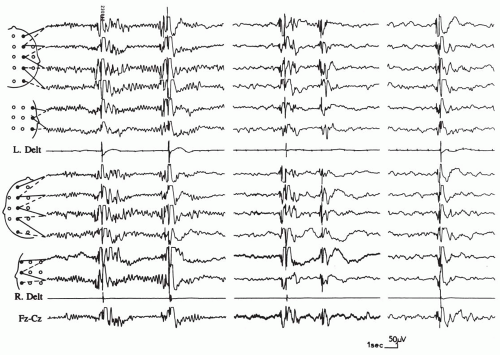 FIG. 6.1. A 4-year-old girl with childhood-onset myoclonic epilepsy having the characteristics of benign myoclonic epilepsy. Isolated generalized jerks are recorded while the patient is drowsy (left) and asleep (middle and right). The jerks are accompanied by generalized spike-wave discharges, but they are clinically indistinguishable from sleep myoclonus. This patient is now 17 years of age, and she has been seizure free since the myoclonic jerks were identified at 4 years of age and were treated with valproate until the age of 7 years. |
Epileptic myoclonus should not be confused with other forms of nonepileptic myoclonus. Spinal or segmental myoclonus involves muscle groups supplied by contiguous segments of the spinal cord or brainstem. The EMG burst duration is longer (more than 100 ms); the jerks usually have a rhythmic appearance (frequency of 1 to 3 Hz), and they constantly involve the same segments. Rhythmic distal myoclonus should be differentiated from tremor, which usually presents with an alternating EMG pattern between antagonist muscles and which is not associated with scalp potentials on back-averaged EEG. Fasciculations present as twitches involving a muscle fascicle, and they are usually well differentiated from myoclonic jerks. They are prominent if the motor unit is enlarged, as occurs in motor neuron diseases in which they usually involve different body segments. Myokymia can be confused with focal myoclonic jerks, but it presents as an undulating, irregular movement involving the facial muscles.
Nosologic Problems
The term myoclonic epilepsies has often been used as a collective designation for a large group of epilepsies characterized by repeated brief seizures that are often responsible for multiple falls, by a severe course that is often resistant to antiepileptic drugs, and by their usual association with mental retardation (Aicardi, 1982a, 1991a). As a result, confusion has arisen because this wide group clearly includes several syndromes with different seizure types that are only superficially similar (Arzimanoglou, 2001).
 FIG. 6.2. Polygraphic electroencephalographic-electromyographic recording of a 4-year-old boy with staggering gait and continuous postural lapses due to epileptic-negative myoclonus. Electromyographic silent periods lasting up to 300 ms appear irregularly, interrupting the ongoing interferential activity recorded from the right deltoid (R. Delt). Silent periods are time locked with sharp waves or spike-wave complexes recorded from the contralateral central area and from the vertex. |
Part of the difficulty arises because several types of brief seizures are manifested by sudden, brief jerks. Although these seizures are superficially similar and, as a consequence, they are often loosely termed myoclonic, they have different neurophysiologic mechanisms. Precise analysis of the ictal manifestations by combined clinical, EEG, EMG, and video monitoring permits the distinction of three different types of seizures. “True” myoclonic seizures are manifested on the EMG by biphasic or polyphasic potentials of 20 to 150 ms in duration that may be followed by a tonic contraction of the affected muscles or a transient suppression of normal tonic activity that lasts up to 400 ms; they may therefore be termed myoatonic seizures (Niedermeyer et al., 1979b; Gastaut and Tassinari, 1975a). On EEG tracings, myoclonic seizures are associated with generalized bursts of polyspike-waves or spike-waves. During atonic seizures, the patient may fall to the ground suddenly or may slump in a rhythmic step-by-step fashion (Erba and Browne, 1983) or he or she may display only brief nodding of the head or sagging of the body. Atonic seizures are accompanied by slow spike-waves on EEG recordings (Lombroso and Erba, 1982; Gastaut et al., 1966a, 1974a), 3-Hz spike-waves (Aicardi and Chevrie, 1971), polyspike-waves (Chayasirisobhon and Rodin, 1981; Gastaut et al., 1974a), or fast recruiting rhythms (Chayasirisobhon and Rodin, 1981; Fariello et al., 1974). The EMG demonstrates the suppression of normal tonic activity in the involved muscles (Gastaut et al., 1974a; Gastaut and Regis, 1961). Tonic seizures involve the tonic contraction of certain muscle groups without a progression to a clonic phase (Erba and Browne, 1983; Gastaut and Broughton, 1972). They can also cause the patient to fall to the ground when the lower limbs
are forcibly flexed or the patient is thrown out of balance, especially if the tonic contraction is asymmetric. Patients who fall in a rigid posture like a statue are having a tonic seizure; those patients who fall by collapsing may be having either an atonic seizure or a global tonic seizure with triple flexion of the lower extremities (Erba and Browne, 1983). During a tonic seizure, the EMG shows an interferential muscle discharge that is similar to that seen with a voluntary contraction. The EEG may show a simple flattening of all activity throughout the attack, a very fast activity (20 ± 5 Hz) of increasing amplitude, or a discharge of a 10-Hz rhythm with a high amplitude from the onset that otherwise is similar to the “epileptic recruiting rhythm” (Brenner and Atkinson, 1982; Fariello et al., 1974; Gastaut et al., 1963, 1974a) (Fig. 4.2). Spasms that, in fact, are quite similar to brief tonic seizures can also occur, and they are an important cause of sudden falls (Egli et al., 1985).
are forcibly flexed or the patient is thrown out of balance, especially if the tonic contraction is asymmetric. Patients who fall in a rigid posture like a statue are having a tonic seizure; those patients who fall by collapsing may be having either an atonic seizure or a global tonic seizure with triple flexion of the lower extremities (Erba and Browne, 1983). During a tonic seizure, the EMG shows an interferential muscle discharge that is similar to that seen with a voluntary contraction. The EEG may show a simple flattening of all activity throughout the attack, a very fast activity (20 ± 5 Hz) of increasing amplitude, or a discharge of a 10-Hz rhythm with a high amplitude from the onset that otherwise is similar to the “epileptic recruiting rhythm” (Brenner and Atkinson, 1982; Fariello et al., 1974; Gastaut et al., 1963, 1974a) (Fig. 4.2). Spasms that, in fact, are quite similar to brief tonic seizures can also occur, and they are an important cause of sudden falls (Egli et al., 1985).
Conversely, myoclonic phenomena, whether massive, bilaterally symmetric, or fragmentary, can occur with different types of epilepsy at different ages (Aicardi, 1986a), and these do not define a single epileptic syndrome. Several types may occur in the same patient and even within the same attack.
The interictal EEG patterns associated with the seizure types described earlier comprise the following two main types: (a) the diffuse slow (1 to 2.5 Hz) spike-wave pattern (see “Clinical and Electroencephalographic Features Common to the ‘True’ Myoclonic Epilepsies”) and (b) the fast (2.5 to 3.5 Hz) irregular spike-wave pattern. Only one of these patterns may be present, or they may be combined and variably distributed. Other EEG abnormalities may also be present. In some patients, especially those with fast spike-wave ictal pattern, the interictal tracings may be normal.
Atonic and tonic seizures are especially common in the epilepsies with brief repeated seizures beginning in children younger than 5 to 6 years. They are much less common in older children, in whom true myoclonic seizures are more common. Thus, separating study of the seizures by the various age-groups is justified.
In infants and children in whom the onset of epilepsy occurs at less than 5 to 6 years of age, several types of seizures, including tonic or atonic types, are usually present in the same patient, and the epileptic syndromes that are encountered are often associated with structural brain damage. The bestknown subgroup in this category is LGS.
In older patients, true myoclonic seizures are the predominant form of attack. Organic brain damage is seldom found; genetic factors are often of etiologic significance; and the prognosis is better, especially for cognitive development. These “true myoclonic epilepsies” are the subject of this chapter.
Even with this approach, some disagreement still exists over the degree of subdivision that is desirable and the limits of accepted syndromes; furthermore, a fair proportion of cases resist precise classification because they display features that are common to different subsyndromes. Therefore, a complete separation of the various syndromes is not possible in all children (Fig. 6.3).
Clinical and Electroencephalographic Features Common to the “True” Myoclonic Epilepsies
The abrupt brief myoclonic seizures usually are repeated many times a day. Like atonic seizures, with which they may sometimes be associated, they can produce only a slight head nodding, often with abduction of the arms, or they may be responsible for falls when the lower limbs are also involved either in the jerk or by the atonic phenomenon that immediately follows the jerk (Tassinari et al., 1992b; Erba and Browne, 1983). A diagnostically useful feature of myoclonic jerks is their short duration (less than 100 ms) and frequent saccadic character, with a rate of repetition at 2 to 3 Hz, which contrasts with the more sustained muscle contraction of most tonic seizures, however brief, and with the slumping of pure atonic attacks. Myoclonic seizures may affect the external ocular muscles and/or the eyelids or facial, especially perioral, muscles in a significant proportion of patients, but the axial type is the most common (57% and 89%, respectively, of 90 patients) (Aicardi and Chevrie, 1971).
Most myoclonic seizures are spontaneous, and they tend to occur at specific times of the day relative to the level of arousal. They occur predominantly on awakening in juvenile myoclonic epilepsy (JME) or while the child is drowsy in benign myoclonic epilepsy (BME). Some jerks may be precipitated by photic stimulation, most commonly in the EEG laboratory, but this may also be seen with natural stimuli (see Chapter 17). Tapping or sudden acoustic stimuli may also cause generalized epileptic myoclonic jerks in infants (Ricci et al., 1995). At times, myoclonic seizures supervene in prolonged series. In some cases, myoclonic status is observed, usually with partial preservation of consciousness (Doose, 1992; Dravet et al., 1992a; Giovanardi Rossi et al., 1991).
The spike-wave activity is usually fast (more than 2.5 Hz). However, occasional myoclonic jerks are associated with slower (2 to 2.5 Hz) discharges. The interictal tracings may be entirely normal or abnormally slow or asymmetric, or they may display focal abnormalities, depending on whether the myoclonic attacks occur as an idiopathic or a symptomatic phenomenon (Aicardi, 1980b). Interictal bursts of irregular polyspike-waves of short duration (less than three per second) often occur spontaneously, or they are induced by photic stimulation (Aicardi and Chevrie, 1971; Dalla Bernardina et al., 1982, 1992b; Dravet et al., 1992a). During non-rapid eye movement (REM) sleep, an increase in the frequency of discharges that generally feature multiple spikes and waves is often seen (Dravet and Bureau, 2002; Dravet et al., 1992a; Aicardi, 1980a; Aicardi and Chevrie, 1971).
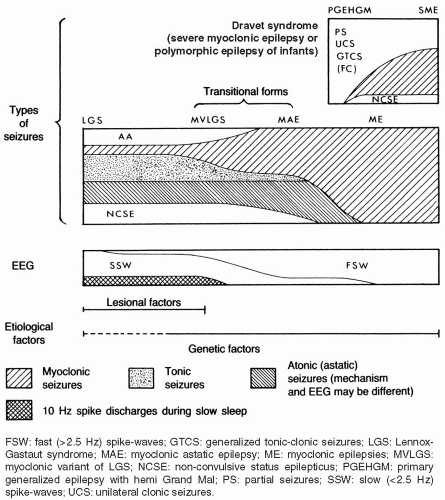 FIG. 6.3. Nosologic concept of the myoclonic epilepsies and Lennox-Gastaut syndrome (LGS). LGS and benign myoclonic epilepsy (BME) are the ends of a clinical spectrum that also includes the myoclonic variant of the LGS (MVLGS), as described by Chevrie and Aicardi (1971); myoclonic-astatic epilepsy (MAE), as defined by Dravet et al. (1982); and other unclassified types of myoclonic epilepsy. Doose’s concept of MAE is based on its supposed genetic etiology. It includes both the severe and the benign idiopathic myoclonic epilepsies. Dravet syndrome (previously termed severe myoclonic epilepsy [SME] or polymorphic epilepsy of infants) belongs to a clinically different spectrum. It begins in the first 2 years of life with convulsive seizures (often febrile), usually followed later by myoclonic and partial seizures. Myoclonic seizures may not be prominent in some closely related cases that feature generalized and/or unilateral tonic-clonic seizures, sometimes with partial attacks. The term primary generalized epilepsy with hemi grand mal has been used for such cases (Doose, 1985). Tonic and atonic seizures and atypical absences in association with interictal diffuse slow spike wave discharges predominate in LGS, while mostly myoclonic discharges associated with fast (>2.5 Hz) spike wave activity characterize the other end. Lesional damage may be predominant in cases of LGS, whereas genetic factors are most important in the myoclonic epilepsies. |
Myoclonic attacks are the only type of seizure in a minority of patients (Dravet et al., 1992b; Aicardi, 1980a). More commonly, they are associated with other seizure types. Generalized tonic-clonic seizures (GTCSs) are most common (Erba and Browne, 1983; Aicardi and Chevrie, 1971), but generalized purely clonic seizures, unilateral clonic attacks, atypical absences, and especially atonic seizures are also observed (Dravet et al., 1992a; Dalla Bernardina et al., 1987). Tonic seizures are not usually seen in predominantly myoclonic epilepsies, but isolated tonic or short tonic-clonic attacks during sleep are not rare in children within the subgroup defined as myoclonic astatic epilepsy (MAE) (Oguni et al., 2002; Guerrini et al., 2002f) (see “Myoclonic Astatic Epilepsy”).
Etiology of the Myoclonic Epilepsies
Symptomatic cases of myoclonic epilepsy are rather uncommon; most often, they are the result of prenatal or perinatal hypoxic-ischemic encephalopathies. Patients with brain damage start having myoclonic seizures between a few months and 2 to 3 years of age (Guerrini et al., 2002b; Elia et al.1998; Dalla Bernardina et al., 1992b; Aicardi, 1980a, 1980b, 1982a). They have developmental delay and clinical and/or imaging evidence of diffuse brain damage. Other seizure types are usually associated with the myoclonias. Myoclonic status may be prominent (Dalla Bernardina et al., 1992b, 2002; Guerrini et al., 1996b; Lombroso and Erba, 1982). The latter authors emphasized the occurrence of continuous mild rhythmic jerks that are hardly detectable without polygraphic recordings and the common association with dystonic movements during wakefulness. Most of their patients were later diagnosed as having Angelman syndrome. The mental outcome was generally poor, even though the myoclonus could sometimes be controlled.
A vast majority of cases are idiopathic or cryptogenic, and genetic factors play an important role in their origins, as indicated by the frequency of a family history of epilepsy in individual forms. The recent observation of mutations of the SCN1A, SCN1B, and GABRG2 genes in some patients with a phenotype said to be consistent with MAE who belong to families with the “generalized epilepsy with febrile seizures plus” (GEFS+) spectrum (Meisler et al., 2001), and of the GABRA1 gene in a family with JME (Cossette et al., 2002) suggests that ion channel disorders may comprise the pathophysiologic substrate for many cases of myoclonic epilepsy.
However, simple inheritance is limited to a few families, and more complex mechanisms, possibly appearing in combination with other factors, are likely in most. The overall incidence of convulsions or epilepsy in first-degree and second-degree relatives of propositi with myoclonic epilepsy has been found to vary from 26% to 38% (Dravet et al., 1992a; Aicardi, 1991a; Giovanardi Rossi et al., 1991), an incidence that is higher than that seen in most other forms of early onset epilepsy and is similar to that in patients with febrile convulsions. A number of authors (Doose, 1992; Doose and Baier, 1987; Doose et al., 1984) have found a positive family history in 37% (16% in siblings) of their cases of “MAE,” which probably includes most cases of cryptogenic myoclonic epilepsy.
Little information, however, is available regarding the pathologic substrate, if any, of the myoclonic epilepsies. Renier and Renkawek (1990) found typical images of microdysgenesis in a biopsy specimen that were similar to those described by Meencke and Janz (1984, 1985) in cases of idiopathic generalized epilepsy (IGE). The significance of such subtle abnormalities has been disputed (Lyon and Gastaut, 1985), and microdysgenetic changes were not observed in a recent study of the brains of five patients with IGE (Opeskin et al., 2000).
SYNDROMES OF MYOCLONIC EPILEPSY IN INFANCY AND EARLY CHILDHOOD
The clinical and EEG features, the etiologic factors, the developmental profile, and the type of seizures that are associated with the myoclonic attacks permit the delineation of some major syndromic groups. In a scheme that has been accepted by several investigators and that was incorporated into the International Classification of Epilepsies, Dravet et al. (1982) recognized the following three major syndromes: BME, severe myoclonic epilepsy (see Chapter 5), and MAE.
Other classifications have been proposed (Lombroso, 1990; Aicardi and Gomez, 1989; Giovanardi Rossi et al., 1988a). Giovanardi Rossi et al. (1988a) proposed the development of an “intermediate” form between the idiopathic and symptomatic epilepsies. Lombroso (1990) held that no firm basis exists for individualizing these two forms and that intermediate types that form a continuum of myoclonic epilepsies, which is somewhat arbitrarily dichotomized, do exist. However, recent findings challenge the view that all forms of myoclonic epilepsy belong to a single large category. Moreover, the recognition of the main syndromic groups does not imply that all cases of myoclonic epilepsies within the same age range must be forcefully classified.
Other investigators (Aicardi and Gomes, 1989) proposed classifying the myoclonic epilepsies simply according to the presence or absence of and the types of associated seizures.
No scheme is really satisfactory, and the differences among the “true” myoclonic syndromes are likely due to the difficulties, or perhaps the impossibility, in assigning precise criteria and limits to the different syndromes proposed.
Benign Myoclonic Epilepsy
The subgroup designated as BME includes cases presenting exclusively or primarily with myoclonic seizures that usually are repeated many times daily. The myoclonic attacks may have been preceded by febrile convulsions. The jerks are preponderantly axial or are generalized; they are usually symmetric. They may result in nodding; staggering; or, only rarely, in falls. Myoclonic seizures in infants, either alone or in combination with subsequent GTCSs do not define, per se, a homogeneous subgroup because idiopathic, cryptogenic, or symptomatic cases may present with similar features (Guerrini et al., 1994a) and these had been classified for many years under the confusing heading myoclonic epilepsies (Jeavons, 1977; Loiseau et al.; 1974; Harper, 1968).
Dravet et al. (1992a) and Dravet and Bureau (1981) initially classified as having BME only those children who had an age at onset of younger than 2 years and who had no other seizure types, except, eventually, febrile seizures. BME was understood to be an early and minor expression of IGE. Similar cases were subsequently recognized by other groups (Ohtsuka et al., 1993; Todt and Muller, 1992; Salas-Puig et al., 1990a; Dalla Bernardina et al., 1983a); a special subgroup, represented by cases of touch myoclonic epilepsy in infants (see Chapter 17) in whom myoclonic jerks are triggered by tactile or sudden acoustic stimuli, was also recognized and may or may not be associated with spontaneous jerking (Ricci et al., 1995; Deonna and Despland, 1989; Revol et al., 1989). The total number of cases described initially remained low, and about half of the patients described by Dravet et al. (1992b) were mildly retarded or behaviorally disturbed at follow-up, casting some doubts on the benign outcome of the syndrome (Aicardi, 1994). Aicardi and Gomes (1988), studying a series of infants and young children presenting with only myoclonic seizures and occasional GTCSs, observed a relatively favorable outcome. However, about half their patients exhibited behavioral and/or learning problems at school age. Therefore, when cases with only myoclonic seizures of early onset were not identified retrospectively, they could apparently have a variable prognosis.
In a recent review of the 103 cases reported thus far under the heading “BME,” Dravet and Bureau (2002) updated their description of the syndrome and slightly modified the initial concepts. They acknowledged that the age at onset may be as late as 5 years (Giovanardi Rossi et al., 1997; Guerrini et al., 1994a), that the term benign is questionable according to the most recent International League Against Epilepsy (ILAE) definitions (Engel, 2001), and that the difference from the milder cases of MAE may not be entirely clear.
For the sake of simplicity, in this section, the authors will continue to use the accepted term BME, bearing in mind the aforementioned uncertainty about its appropriateness.
Etiologic and Epidemiologic Data
BME is rare. Dravet and Bureau (2002) reported that children with such a syndrome represented less than 1% of the epilepsy population in their specialized center and 2% of all IGEs. Other authors observed BME in 1.3% to 1.7% of the children with seizure onset in the first year of life (Sarisjulis et al., 2000; Caraballo et al., 1997) and in 2% of those with onset in the first 3 years. The syndrome is almost twice as common in boys than in girls (Dravet and Bureau, 2002).
A family history of epilepsy or febrile seizures is observed in almost 40% of children with BME (Dravet and Bureau, 2002; Aicardi and Gomes, 1988). However, familial cases have not been described. Occurrence of MAE in the brother of an affected child has been reported (Arzimanoglou et al., 1996).
Clinical and Electroencephalographic Presentation
With very few exceptions, the child’s development is normal before the first myoclonic seizures appear. The age at onset ranges from 5 months to 5 years. The initial
jerks tend to be mild and rare, and they are often overlooked or misinterpreted as spasms or tics. They are usually manifested as head drops or upward eyeball rolling that is accompanied by a brisk abduction of the upper limbs (Dravet and Bureau, 2002). Myoclonic jerks occur several times daily, sometimes clustering within a short time, and they may vary in intensity in the same child. Stronger jerking that is accompanied by projection of objects or falling are rare, and it tends to appear later in the course, if at all. Rhythmic or quasi-rhythmic repetition of a short series of jerks within 5 to 10 seconds is another feature usually seen in older children.
jerks tend to be mild and rare, and they are often overlooked or misinterpreted as spasms or tics. They are usually manifested as head drops or upward eyeball rolling that is accompanied by a brisk abduction of the upper limbs (Dravet and Bureau, 2002). Myoclonic jerks occur several times daily, sometimes clustering within a short time, and they may vary in intensity in the same child. Stronger jerking that is accompanied by projection of objects or falling are rare, and it tends to appear later in the course, if at all. Rhythmic or quasi-rhythmic repetition of a short series of jerks within 5 to 10 seconds is another feature usually seen in older children.
If falling occurs, it characteristically is followed by the immediate resumption of the previous posture and ongoing activities, usually without injury. This behavior is extremely different from falling that is linked to an epileptic spasm (i.e., an atonic seizure), which clearly interferes with ongoing activity as the children appear confused and often cry because they are frightened or they have suffered injuries. Mild myoclonic jerks cause little displacement of the limbs, and their expression is influenced by the child’s posture. An occasional myoclonic seizure may be asymmetric or even unilateral. Whether impairment of consciousness occurs is difficult to appreciate because the jerks are too brief and, in general, no detectable interruption of ongoing activities occurs; however, repeated jerks might be accompanied by a mild impairment of contact. Drowsiness appears to facilitate the jerks. Episodes of myoclonic status are extremely rare (Aicardi and Gomes, 1988).
A subset of children presents with generalized myoclonic jerks that are triggered by sudden tapping or acoustic stimuli, and these may or may not be associated with spontaneous attacks of an identical type (Ricci et al., 1995; Deonna and Despland, 1989; Revol et al., 1989). The electroclinical characteristics of this form are similar to those seen in children with only spontaneous jerks, and its evolution might be even more “benign” (Dravet and Bureau, 2002). The onset of reflex jerks may be as early as 4 months of age (Ricci et al., 1995). These jerks can be provoked while the child is awake or during sleep.
The EEG shows normal background activity in almost all children with BME (Dravet and Bureau, 2002; Guerrini et al., 1994a; Aicardi and Gomes, 1988). Interictal EEG, especially while the child is awake, may be normal because spike-wave discharges very rarely occur without concomitant myoclonic jerks. Focal EEG abnormalities have been reported occasionally (Dravet and Bureau, 2002; Lin et al., 1998; Giovanardi Rossi et al., 1997), but, in most patients, they disappeared in follow-up recordings. Sleep recordings provide higher chances of capturing ictal discharges, which are represented by bursts of generalized fast spike-waves or polyspike-waves without rhythmicity, and of brief duration (less than 3 seconds). Most jerks are isolated and time locked with the spike component of the spike-wave complex (Guerrini et al., 2002b). In the reflex variety of BME, a refractory period lasting up to 2 minutes may be observed after a reflex-induced jerk (Ricci et al., 1995). In about 10% of all affected children, myoclonic jerks are also triggered by light stimuli, especially intermittent photic stimulation (IPS) in the EEG laboratory (Dravet and Bureau, 2002; Guerrini et al., 1994a; Aicardi and Gomes, 1988).
Course and Outcome
The course of BME is said to be benign, an opinion largely based on retrospective studies that mainly included children selected because of their favorable outcomes. In some series (Dravet and Bureau, 2002), one of the proposed criteria for diagnosis was a rapid response to valproate monotherapy, which is clearly also a strong criterion of benignity. However, some children did have cognitive or behavioral sequelae, indicating that, even with these strict criteria, a benign outcome could not be guaranteed.
The epilepsy outcome is said to be favorable. Myoclonic jerks were reported to have disappeared in all the children who were followed for a long period (Dravet and Bureau, 2002). Dravet and Bureau (2002) estimated that myoclonic seizures had been present for less than 1 year in most of the 52 published cases for whom this information was available. However, the mean delay for initiating an effective treatment was not known. In general, treatment had been withdrawn in most patients older than 6 years at follow-up. A minority of patients were reported to have experienced rare GTCSs when they were 9 to 16 years of age (Dravet and Bureau, 2002; Giovanardi Rossi et al., 1997; Dravet et al., 1992b). In some, these seizures occurred during valproate withdrawal, and they were subsequently controlled by the reinstitution of treatment (Dravet and Bureau, 2002). No detailed information is available for the remaining patients. The main cause for continuing treatment after that age was photosensitivity that had either persisted or had emerged after the spontaneous myoclonic jerks disappeared (Dravet and Bureau, 2002; Lin et al., 1998; Giovanardi Rossi et al., 1997).
Some children with BME did not receive any drug treatment (Dravet and Bureau, 2002). Although no additional seizure types were seen, the myoclonic jerks had persisted. These cases were thought to have
an increased risk of impaired psychomotor development and behavioral disturbances, but evidence for this was unconvincing.
an increased risk of impaired psychomotor development and behavioral disturbances, but evidence for this was unconvincing.
A critical review of the published information on treatment indicated that most children were well controlled with valproate monotherapy and that they were seizure free by school age (Dravet and Bureau, 2002). Other drugs, including phenobarbitone or nitrazepam monotherapy or combinations of valproate or phenobarbitone with clonazepam, clobazam (Giovanardi Rossi et al., 1997), or ethosuximide (Todt and Muller, 1992), have been used, especially in the past.
The cognitive and behavioral outcome is favorable (Dravet and Bureau, 2002). Only a minority (17%) of affected children experienced mild cognitive difficulties. Clearly, the good outcome of BME fulfilling the criteria of Dravet and Bureau (2002) might be due to the additional requirements for criteria of benignity, such as a rapid response to therapy, rather than to an intrinsic difference in the severity of the condition. Similar remarks could apply to those cases classified as “MAE” with a favorable outcome (Oguni et al., 2002; Kaminska et al., 1999). Thus, BME does not always have a benign outcome, nor is it the only relatively benign syndrome among the myoclonic epilepsies.
Diagnostic and Nosologic Aspects
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


