For an up-to-date review of an individual metabolic disease or to find a lab where an analyte or gene test can be sent, we recommend visiting the NIH GeneReviews site at http://www.genetests.org.
METABOLIC DISORDERS IN THE NEWBORN AND YOUNG INFANT
Disorders of Neurotransmitter Synthesis and Removal
Tetrahydrobiopterin and Guanine Triphosphate Cyclohydrolase Deficiency
Tetrahydrobiopterin (BH4) is a cofactor for the enzymes involved in converting tyrosine and tryptophan to levodopa and serotonin. Deficiencies in BH4 lead to disruptions in the neurotransmitter amines, levodopa, and serotonin. It is one of several disorders of neurotransmitter production. Defects most commonly occur due to abnormalities of guanine triphosphate cyclohydrolase (GTPCH), the first enzyme involved in the multistep process of converting guanine triphosphate to BH4. However, defects in other enzymes in the pathway have also been identified.
GCH1 is the gene involved in GTPCH enzyme formation. Autosomal dominant mutations lead to early onset dopa-responsive dystonia (Segawa disease), and autosomal recessive mutations lead to a neonatal onset encephalopathy. Genetic testing for GCH1 mutations and several other enzyme deficiencies in the BH4 synthesis pathway is available as are gene panels to screen for mutations in many of the genes involved in neurotransmitter synthesis.
Symptoms typically involve variable and fluctuating levels of psychomotor retardation, convulsions, microcephaly, swallowing difficulties, truncal hypotonia, limb hypertonia, involuntary movements, and oculogyric crises. Some of these symptoms begin shortly after infancy. A diurnal pattern of worsening of symptoms may be described. Of clinical note, other enzyme abnormalities both before and after BH4 production (including α-amino decarboxylase [AADC] deficiency involved in converting levodopa to active dopamine) can lead to identical symptoms (1).
Diagnosis of many of the neurotransmitter disorders is typically made by measuring levels of spinal fluid neurotransmitter metabolites (5-hydroxyindoleacetic acid and homovanillic acid) and precursors (biopterin, neopterin, and 3-methyl-dopa). Elevations in plasma and cerebrospinal fluid (CSF) phenylalanine may be seen in the amino acid profile of some patients, but this finding is frequently not present (1). Urine pterin concentrations can aid in differentiation between the various defects of biopterin synthesis, though this testing is not readily available in North America at this time.
Treatment includes tetrahydrobiopterin supplementation, pharmacotherapy with levodopa–carbidopa, and 5-hydroxytryptophan. Pyridoxine is an important cofactor, and supplementation may benefit patients. In AADC deficiency a dopamine agonist and monoamine oxidase inhibitor are used. Treatment is beneficial to varying degrees (2).
Glycine Encephalopathy (Formerly Nonketotic Hyperglycinemia)
In this autosomal recessive inborn error of amino acid metabolism, large amounts of glycine accumulate in the body, especially the brain, because of a defect in the multienzyme complex for glycine cleavage. The enzyme system is confined to the mitochondria and is composed of four protein components (designated P, H, T, and L), three of which have a gene identified. GLDC, localized to 9p22, is the most common gene affected, with mutations leading to abnormal functioning of the P protein (3).
The pathophysiology of nonketotic hyperglycinemia (NKH) has not been fully elucidated, but the elevated glycine is believed to impact the central nervous system (CNS) via its role as an inhibitory transmitter in the brainstem and spinal cord, and an excitatory transmitter in the cortex. The majority of cases present within the first 48 hours of life with lethargy, respiratory difficulties, apnea, and seizures that are often myoclonic or characterized as infantile spasms. Cortical malformations or corpus callosum defects may be present. A burst-suppression EEG pattern with variable intervals between the bursts alternating with epochs of greater continuity is characteristic. The combination of early-onset seizures with prominent myoclonus and variable burst-suppression EEG is also called EME, as described by Aicardi. Glycine encephalopathy is an important cause of EME evolving to hypsarrhythmia, although this is more commonly observed in Ohtahara syndrome. Ohtahara syndrome is thought by some authorities to be more commonly associated with structural abnormalities and not as highly associated with errors of metabolism (1).
Later-onset forms of this disease have also been described, with patients having varying degrees of epilepsy, retardation, and movement abnormalities. An adolescent/adult-onset form presents with associated spastic paraparesis and optic atrophy (4).
Laboratory testing reveals elevations of glycine in the plasma, urine, and CSF amino acid profile. The absence of excess ketones in the blood and urine, and of abnormal organic acids in the urine, helps to differentiate NKH from other conditions associated with hyperglycinemia, such as propionic and methylmalonic acidemias. The ratio of CSF to serum glycine is also helpful, as it is significantly elevated in patients with NKH. Genetic sequencing for the three known loci is clinically available (5).
There is no effective long-term treatment for this disorder. High doses of benzoate may reduce CSF glycine and improve seizure control, but it does not appear to stop the development of mental retardation (6,7). Because benzoate treatment may deplete carnitine levels, carnitine supplementation is recommended when benzoate is used (8). Dextromethorphan, an NMDA antagonist, has been tried for this condition as well. Valproic acid should be avoided because it induces hyperglycinemia. Strychnine, a glycine antagonist, and diazepam have been reported to blunt seizures, but have not influenced the long-term outcome (9). Prognosis is poor, with progressive microcephaly and intractable epilepsy. Even when treated, death often occurs within the first few months to years of life (10).
A transient form of NKH exists with similar early clinical and biochemical findings. In this condition, glycine concentrations normalize between 2 and 8 weeks of life, and prognosis is favorable (11).
Serine and 3-Phosphoglycerate Dehydrogenase Deficiency
3-Phosphoglycerate dehydrogenase (PHGDH) deficiency is an autosomal recessive condition that results in impaired L-serine biosynthesis due to a mutation in the PHGDH gene (1p12). Serine is the precursor of D-serine and glycine, both potent neurotransmitters. Serine also has a role in myelin production (12).
Typically there is a pre- or perinatal onset of symptoms including congenital microcephaly, intractable seizures, spastic quadriparesis, and profound cognitive delays. The magnetic resonance imaging (MRI) may show white matter lesions. The condition is often misdiagnosed as cerebral palsy. Reduced serine levels are found in CSF amino acid specimens though glycine levels are not always low. Plasma serine and glycine levels may only be low in fasting specimens but are not reliable indicators as they may also be normal (13).
Enzyme activity can be measured in skin fibroblasts. Genetic testing is clinically available. Treatment involves supportive care and L-serine supplementation (14).
Succinic Semialdehyde Dehydrogenase Deficiency
Succinic semialdehyde dehydrogenase (SSADH) deficiency is an autosomal recessive disorder impairing γ-aminobutyric acid (GABA) catabolism. The enzyme is involved in the second and final step of GABA degradation. GABA, an inhibitory neurotransmitter, accumulates (15). The ALDH5A1 (6p22) gene encodes this enzyme, and mutations here account for over 97% of cases (16).
The disorder is slowly progressive with infancy to childhood onset. Seizures occur in more than 50% of patients. Other symptoms include varying degrees of ataxia, hypotonia, speech delay, and mental retardation. A movement disorder with hyperkinetic movements, choreoathetosis, dystonia, and myoclonus can be seen with earlier onset, and presenting as a more severe form of this disease. Aggressive behaviors and self-injury may also occur (17).
Diagnosis is initially made by finding elevations of 4-hydroxybutyric (4HB) acid in urine organic acids, plasma, or CSF. Of clinical note, the 4HB peak can be easily missed on routine organic acid analysis due to its coeluting with a large urea peak. Thus, one must ensure that the lab processing the specimen is monitoring for this ion. Variable elevations of glycine can be seen in the amino acid profile. Confirmatory gene sequencing is clinically available. The EEG may show generalized background slowing and spike discharges (18).
Treatment of this disorder is symptom based. Vigabatrin, an irreversible inhibitor of GABA transaminase, has been used in some individuals with positive responses (including increased socialization, behavioral improvement, increased alertness, and reduced ataxia), but treatment with this agent has worsened symptoms in others. Valproate use is contraindicated as it may inhibit residual SSADH enzyme activity (19–21).
Vitamin and Mineral Metabolism–Related Diseases
Pyridoxine- (and Folinic Acid–) Responsive Epilepsy
In pyridoxine-responsive epilepsy, refractory seizures typically develop within the first several days of life. These may be characterized by infantile spasms or have a variety of partial, myoclonic, and atonic features. Atypical presentations include a later onset of seizures, seizures that initially respond to treatment and then become intractable, and seizures with only a partial initial response to pyridoxine.
The disorder occurs due to a mutation in the ALDH7A1 gene (5q31). The ALDH7A1 gene encodes for the protein antiquitin, which is involved in lysine catabolism. While it is not directly involved in pyridoxine metabolism, disruptions of antiquitin function secondarily deplete pyridoxine (22). Pyridoxine plays several critical roles in the brain, including the conversion of levodopa to dopamine and glutamate inactivation via glutamic acid decarboxylase.
Diagnosis has typically been made clinically, with seizures abating after a high dose of intravenous (IV) pyridoxine (100 to 500 mg). The initial EEG is characterized by generalized bursts of high-voltage delta activity interspersed with spike and sharp waves and periods of asynchronous attenuation. After treatment there is conversion of the EEG to a burst-suppression pattern, and later normalization with subsequent doses. This change occurs within minutes and may persist for hours, or even a day (23). Confirmatory testing is via spinal fluid analysis, as described below. Testing for ALDH7A1 mutations is clinically available.
A clinical and genetic link has been made between folinic acid–responsive epilepsy, pyridoxine-responsive epilepsy, and ALDH7A1 mutations, thus leading one to conclude that folinic acid– and pyridoxine-responsive seizures are one and the same. Previously it was believed that folinic acid–responsive epilepsy was a rare yet separate metabolic epileptic encephalopathy with a diagnosis made by identifying a characteristic peak of a yet unknown compound in spinal fluid neurotransmitter analysis (24).
More recently, a patient with folinic acid–responsive epilepsy was found to respond to pyridoxine supplementation (25). Eventually, individuals with folinic acid–responsive epilepsy were identified as having ALDH7A1 mutations and patients identified as having pyridoxine-dependent epilepsy (mutation confirmed cases) were found to have the characteristic folinic acid–responsive epilepsy peak identified in their CSF (26).
In evaluating these patients, plasma, urine, and spinal fluid levels of pipecolic acid may also be elevated though this chemical is not a sensitive biomarker (27). A link also exists to elevated urinary and CSF concentration of α-aminoadipic semialdehyde (α-AASA), and clinical testing for this compound is available. The characteristic peak of this yet unidentified compound is found when spinal fluid neurotransmitter testing is run. Spinal fluid folate levels are normal in this condition. As the link between folinic acid– and pyridoxine-responsive epilepsy is secure, routine CSF analysis for diagnosis is crucial. Early treatment is also critical, as it improves developmental outcome. Maintenance therapy with pyridoxine and folinic acid is necessary. Onset of disease may extend beyond the newborn period, making this disorder a consideration in older infants with refractory seizures as well (28,29). Due to pyridoxine and folic acid’s relatively decreased ability to cross the blood–brain barrier, treatment with pyridoxal 5-phosphate and folinic acid is recommended.
Pyridoxal-L-Phosphate-Responsive Epilepsy
Some neonates and children with an epileptic encephalopathy seemed to respond to pyridoxal-L-phosphate (PLP) supplementation as opposed to pyridoxine. Further research showed that some of these individuals had a defect in an enzyme pyridoxal/PLP—pyridox(am)ine 5′-phosphate oxidase (PNPO) required for the synthesis of intracellular PLP from dietary pyridoxine. Eventually, the PNPO gene (17q21.2) was identified and these mutations were confirmed as the cause of this relatively rare condition. The clinical presentation in these children is similar to those with pyridoxine-dependent epilepsy and disorders of neurotransmitter metabolism as described above. Treatment, however, requires PLP as opposed to pyridoxine (30). Diagnosis is aided by measuring pyridoxal 5-phosphate levels in spinal fluid.
Molybdenum Cofactor Deficiency and Sulfite Oxidase Deficiency
The rare conditions of molybdenum cofactor deficiency (MOCOD) and sulfite oxidase deficiency present shortly after birth, also with a progressive encephalopathy, feeding difficulties, hypotonia, and refractory partial, myoclonic, or apparently generalized seizures. Dysmorphic features, lens dislocation, and hepatomegaly are all characteristic findings (31). The two conditions have an essentially identical clinical phenotype, likely due to both conditions leading to the loss of sulfite oxidase function. As typical of all of these diseases, later-onset and relatively milder and varying phenotypes have been described in the literature.
Molybdenum cofactor is critically needed for the proper function of three enzymes: sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase. Sulfite oxidase converts sulfite to sulfate. Xanthine dehydrogenase converts xanthine to hypoxanthine to eventually form uric acid. Aldehyde dehydrogenase is involved in the reverse reaction of hypoxanthine to xanthine (32).
MOCOD is due to mutations in one of two genes, MOCS1, at 6p21.3 or MOCS2, at 5q11 (33). Sulfite oxidase deficiency is typically due to mutations in the SUOX gene on chromosome 12 (34).
Patients typically present with a metabolic crisis, developing irritability, opisthotonic posturing, and seizures. The EEG in these patients may show multifocal paroxysms and a burst-suppression pattern. Neuroimaging may show poor differentiation between the gray and white matter, severe cerebral and cerebellar atrophy, and multiple cystic cavities in the white matter. Diagnosis is made by a variety of methods. Since MOCOD and isolated sulfite oxidase deficiency both result in high levels of urinary sulfites, a sulfite dipstick on a fresh urine sample may detect abnormalities, though this test has a high false-negative rate (35,36). Plasma uric acid levels may be low in MOCOD, but not in sulfite oxidase deficiency. Both disorders will lead to an accumulation of urine S-sulfocysteine. The two disorders can be distinguished on laboratory testing as elevations of urine purines and pyrimidines (xanthine and hypoxanthine) occur in MOCOD but not in sulfite oxidase deficiency. The enzyme deficiencies can be demonstrated in cultured fibroblasts and liver tissue. Genetic testing is clinically available. No effective treatment has yet been identified and prognosis is poor, with death occurring within the first days to weeks of life (37). Clinical trials with cyclic pyranopterin monophosphate, an intermediary precursor of molybdenum cofactor, are planned after they showed potential benefit in slowing disease progression in several patients with MOCOD (38).
Cerebral Folate Deficiency
Folate is concentrated in the nervous system, and the CSF concentrations of folate are higher than the serum concentrations. Deficiencies in cerebral folate lead to a slowly progressive encephalopathy, with intractable seizures. If it occurs in conjunction with systemic folate deficiency, megaloblastic anemia, mouth ulceration, diarrhea, and failure to thrive may also be seen. Neuroimaging may reveal calcifications in the occipital lobes and basal ganglia (39).
Reductions in cerebral folate can occur primarily due to a mutation in the gene encoding the folate transporter (FOLR1) or secondarily due to systemic disease, gut malabsorption syndromes such as colitis, primary mitochondrial disorders (40), many other metabolic diseases (see section Methylenetetrahydrofolate Reductase Deficiency), and certain genetic syndromes (Rett syndrome) (41). Most patients with cerebral folate deficiency do not have a defect in the FOLR1 gene (42). This condition is different from folinic acid–responsive epilepsy described above.
There is also an association of lactose autoantibodies cross reacting with and blocking the folate receptor, potentially leading to a secondary cerebral folate deficiency (43). A lactose-free diet has been considered as a treatment in these situations (44). However, testing for folate autoantibodies is not clinically available and this association needs further study.
The disorder is typically diagnosed by finding low levels of methyltetrahydrofolate (MTHF) in CSF. Normal amino and organic acids analysis and normal plasma folate levels help exclude other potentially treatable causes. If another syndromic, metabolic, or systemic cause of low cerebral folate is not identified, specialized testing for folate autoantibodies is available on a research basis (43).
Treatment involves supplementation with IV and oral folate. Folinic acid may be used since it has better blood–brain barrier penetration than does folic acid. If lactose-mediated autoantibodies are identified, a lactose-free diet might be considered (44). Outcome is still poor.
Methylenetetrahydrofolate Reductase Deficiency
Methylenetetrahydrofolate reductase deficiency (1p36.3) is the most common inborn error of folate metabolism (45). The metabolic defect results from insufficient production of 5-MTHF, which is needed for the remethylation of homocysteine to methionine, because of a deficiency in methylenetetrahydrofolate reductase. In affected individuals, a progressive neurologic syndrome develops in infancy. Children with this disorder have acquired microcephaly and seizures characterized by intractable infantile spasms, generalized atonic and myoclonic seizures, and focal motor seizures. EEG findings vary from diffuse slowing of background activity to continuous spike–wave complexes or multifocal spikes. The early-onset form differs from the late-onset form. The latter presents with progressive motor deterioration, schizophrenia-like psychiatric symptoms, and recurrent strokes; seizures are uncommon. Homocystinuria and elevated serum concentrations of homocysteine with reduced or normal serum methionine are the main biochemical features. These findings can be caused by several other amino acid disorders as well. Dietary supplementation with folic acid, betaine, and methionine has proven beneficial. In the acute setting, high-dose methionine has been effective in stopping seizures (46).
Defects in methionine biosynthesis are also associated with seizures. Convulsions are frequent and are predominantly generalized, although myoclonic seizures with hypsarrhythmia have been reported. Diagnostic laboratory findings are megaloblastic anemia, homocystinuria, decreased methionine, and normal folate and cobalamin concentrations in the absence of methylmalonic aciduria (46). These conditions are typically diagnosed via extended newborn screening (NBS) in North America and treated presymptomatically. Thus patients presenting with new-onset epilepsy who have obtained extended NBS are less likely to have this condition.
Inborn Errors of Creatine Metabolism
Creatine represents a storage depot of adenosine triphosphate (ATP) for various tissues including the brain. Depletion of cerebral creatine due to inborn errors in synthesis or transport leads to a progressive encephalopathy and epilepsy. Creatine forms via a two-step enzymatic path, with arginine converted to guanidinoacetate via arginine:glycine amidinotransferase (AGAT) and then to creatine via guanidinoacetate N-methyltransferase (GAMT). Deficiencies in both enzymes have been identified, and the genetic loci for AGAT and GAMT are known. Creatine, once formed, is transported into the cell via a specific transporter, and abnormalities in creatine transport can also occur due to defects in the SLC6A8 gene at Xq28 (47).
Development can be delayed from the beginning or after a regression beginning between 3 months and 2 years of age. Seizures may present in the first months of life with generalized tonic–clonic, astatic, absence, myoclonic, or partial events. Multifocal epileptiform discharges have been reported on the EEGs of affected individuals (48). Other clinical features may include dystonia, dyskinesias, microcephaly, and autistic behaviors (49). A mild form presenting with severe speech delay, mild autism, and infrequent seizures has also been identified (50).
Diagnosis is typically via quantifying urine, plasma, and/or spinal fluid guanidinoacetate and creatine. Low creatinine is not a reliable marker of the condition. Brain magnetic resonance spectroscopy (MRS) shows a reduced creatine peak (51).
Supplementation with creatine monohydrate (350 mg/kg/day to 2 g/kg/day) has led to improvement in affected individuals though not in patients with creatine transporter disorders (47).
Early-Onset Multiple Carboxylase Deficiency (Holocarboxylase Synthetase Deficiency)
Early-onset multiple carboxylase deficiency presents in the first week of life with lethargy, respiratory abnormalities, irritability, poor feeding, and emesis. A skin rash is present in more than 50% of patients. Generalized tonic convulsions, partial motor seizures, and multifocal myoclonic jerks develop in 25% to 50% of cases. This disease is tested for in the neonate in certain states via expanded NBS and can often be treated prior to symptom onset.
A deficiency in the enzyme holocarboxylase synthetase (HLCS) leads to a decrease in holocarboxylase. As this enzyme links biotin to four carboxylases in the mitochondria and one in the cytosol, an inactivity of all carboxylases results. The HLCS gene locus is 21q22.1. Although rare, this condition is very important to recognize because prompt treatment with biotin may result in dramatic improvement. Laboratory findings demonstrate ketoacidosis and a characteristic pattern on organic acid analysis. Hyperammonemia may be seen with acute episodes. Electrographically, a burst-suppression pattern or multifocal spikes are observed. Definitive diagnosis can be made by enzyme assays, and gene sequencing. Treatment with biotin (10 mg/day) produces clinical improvement (52).
Late-Onset Multiple Carboxylase Deficiency (Biotinidase Deficiency)
This disease is also screened for in certain states via expanded NBS. The disorder involves the body’s ability to recycle biotin via biotinidase. The BTD gene has been identified at the 3p25 locus (53).
When not diagnosed early, seizures are a prominent feature occurring in 50% to 75% of affected children. In fact, seizures are the presenting feature in 38% of patients and may be generalized tonic–clonic, partial, myoclonic, or infantile spasms. Symptoms often begin at 3 to 6 months of age, with hypotonia and developmental delay. Seborrheic or atopic dermatitis and alopecia are common. As the disease progresses, ataxia, optic atrophy, and sensorineural hearing loss develop. Later-onset and/or milder phenotypes exist due to partial enzyme deficiency. EEG findings may include a suppression-burst pattern, absence of physiologic sleep patterns, poorly organized and slow waking background activity, and frequent spike and spike-slow-wave discharges (54).
Diagnosis is typically made via abnormalities in urine organic acid and plasma acylcarnitine analysis. Biotinidase enzyme activity can be measured in leukocytes and cultured fibroblasts. Genetic sequencing is clinically available. When this condition is considered as a differential diagnosis, a therapeutic trial with high-dose oral biotin should be started while awaiting test results.
Menkes Disease (Kinky-Hair Disease)
An X-linked disorder of copper absorption, Menkes disease was first described by Menkes et al. in 1962. Defects in the copper transporting ATPase gene (ATP7A, Xq12–q13) impairs intestinal copper absorption, reduces copper export from tissues, and decreases the activity of copper-dependent enzymes, including dopamine β-hydroxylase (55).
A characteristic twisting of the hair shaft, resulting in “kinky hair” of the head and eyebrows, is noted on microscopic examination of the poorly pigmented hairs. Affected boys may be premature and may have neonatal hyperbilirubinemia or hypothermia. Progressive neurologic deterioration with spasticity is present by 3 months of age, and children may have associated bone and urinary tract abnormalities as well. The disease has a rapidly fatal course (1).
Seizures are a prominent feature in Menkes disease, with intractable generalized or focal convulsions. Infantile spasms have also been reported (56). Stimulation-induced myoclonic jerks may be present. Multifocal spike and slow-wave activity can be seen on the EEG, sometimes resembling hypsarrhythmia (57).
Laboratory testing reveals extremely low serum copper and ceruloplasmin levels. Elevations in CSF lactate may be seen, and there is low total copper content in the brain. Neuroimaging may show brain atrophy, focal areas of necrosis, and subdural collections. Brain magnetic resonance angiography (MRA) shows dilated and tortuous intracranial blood vessels. Genetic testing of the ATP7A gene via sequencing and deletion analysis identifies almost all patients with Menkes (55).
There is no fully effective treatment. Daily copper injections may be beneficial if administered early in the course of the disease.
Overlap exists between Menkes disease and occipital horn syndrome (58). It is now known that both Menkes and occipital horn syndrome conditions are allelic due to mutations in the same gene (58).
Disorders of Carbohydrate Metabolism
Glut-1 Transporter Deficiency Syndrome
The Glut-1 transporter deficiency syndrome was first described in 1991 (59). This autosomal dominant condition results from a loss of functional glucose transporters, encoded by the SLC2A1 gene, that mediate glucose transport across the blood–brain barrier (60). Clinical features include developmental delay, ataxia, hypotonia, infantile seizures, and acquired microcephaly. There is a reduction in the CSF-to-blood glucose ratio to half of normal (typically, CSF glucose is <40 mg/dL). In addition, a low lactate concentration might be seen. Additional confirmation of impaired glucose transport can be performed through assays in erythrocytes (61), and clinical genetic testing is available.
Seizures, specifically neonatal ones, are often the first identified feature of this syndrome though patients with later onset and mild epilepsy have been described. Typical seizure types include absence, myoclonic, astatic, generalized tonic–clonic, and partial–complex. About 10% of patients have no clinical seizures. A normal EEG is commonly seen between seizures, although generalized 2.5- to 4-Hz spike–wave discharges are observed in more than one-third of children older than 2 years of age (62).
Affected individuals without early-onset refractory epilepsy have been identified, and a screening for lumbar puncture should be considered in those individuals as well (63). Early initiation of the ketogenic diet is effective in the treatment of seizures as well as overall disease progression, as it provides an alternative cerebral energy source (64).
More recently, patients with Glut-1 diseases presenting with early-onset atypical absence epilepsy and, less frequently, idiopathic generalized epilepsy have been identified (65,66). Some patients may present with isolated movement disorders. Paroxysmal exertional dyskinesia (PED) is now recognized as an allelic variant of Glut-1 deficiency (67,68). Some patients with PED also have epilepsy. In patients without the classic presentation, hypoglycorrhachia may be milder. Genetic testing is often needed for diagnosis (69).
Other Disorders
Fructose 1,6-bisphosphatase deficiency, a rare, potentially life-threatening disorder of gluconeogenesis, presents within the first few days of life with respiratory abnormalities, hypotonia, lethargy, hepatomegaly, irritability, and convulsions. Laboratory findings reveal lactic acidosis, ketosis, hypoglycemia, elevated plasma concentrations of alanine, and the presence of abnormal urinary organic acids with glycerol and glycerol-3-phosphate (70). The FBP1 gene is located at 9q22.2–q22.3 (71). Neurologic sequelae can be prevented by avoidance of hypoglycemia.
Hereditary fructose intolerance (fructose 1,6-biphosphate aldolase deficiency) may be seen in the neonatal period in infants who are formula fed and given fructose or sucrose early in life. Symptoms include profound hypoglycemia, emesis, and convulsions. If the disease is readily diagnosed, fructose and sucrose can be eliminated from the diet before significant cerebral injury occurs (70).
Mitochondrial Disorders
Disorders of energy metabolism typically present with later-onset epilepsy outside of the immediate newborn period. However there are exceptions to the rule, especially when discussing the ever-growing array of mitochondrial phenotypes.
Mitochondria are the cell’s energy factories, though they also have a key role in initiating apoptosis, and reactive oxygen species formation and removal. When not functioning properly, organs most dependent on cellular energy show symptoms—especially the brain. While multi-organ involvement and lactic acidosis were initially described as sine qua nons of the disease, these findings are not reliably present and the vast majority of patients do not present with the classically described syndromes. These phenotypes, such as MELAS, myoclonic epilepsy with ragged-red fibers (MERRF) and at least 10 others, created order to a set of novel and often unrelated symptoms prior to our current knowledge of the disease. We now know that almost any unexplained neurologic symptom can be due to mitochondrial dysfunction, especially refractory epilepsy. The epilepsy may occur in isolation, or with other neurologic problems including optic nerve disease, retinal pigmentary changes, hearing loss, developmental delays, neuropathy, and myopathy. Myoclonic epilepsy has been associated with mitochondrial disease, but patients with almost any seizure type, including generalized epilepsy, are seen (72).
These conditions typically occur due to genetic abnormalities leading to aberrant mitochondrial function. Over 1500 nuclear DNA (nDNA) genes are involved in mitochondrial formation and function, along with maternally inherited mitochondrial DNA (mtDNA) providing an additional 37 genes. Thus, most cases of mitochondrial disease are autosomal recessive in inheritance, and <20% have a maternal mtDNA-based inheritance pattern. Diagnostic testing initially involves looking for a combination of biochemical abnormalities in plasma amino acids, acylcarnitines, lactate, pyruvate, and urine organic acids (73).
While biochemical and pathologic studies of mitochondria in muscle and other tissue are at times needed to provide a diagnosis, advances in genomic medicine has led to the availability of a wide array of next-generation sequencing testing for mitochondrial disease. For some patients, diagnosis is routinely made using DNA testing, circumventing the need for invasive studies.
Treatment varies and includes preventing worsening during metabolic or physiologic stresses, avoiding mitochondrial toxins and poisons, use of select cofactors and supplements, and providing symptomatic care (74).
Pyruvate Dehydrogenase Deficiency
The mitochondrial pyruvate dehydrogenase (PDH) complex is composed of three enzymes: pyruvate decarboxylase (E1), dihydrolipoyl transacetylase (E2), and dihydrolipoyl dehydrogenase (E3). The E1 enzyme is itself a complex structure, a heterotetramer of two α and two β-subunits. The E1 α-subunit is particularly important, as it contains the E1 active site. Mutations in the PDHA1 gene (Xp22.2–p22.1) encoding for this region are the most common cause of PDH deficiency (75).
PDH deficiency has a wide variety of clinical presentations, ranging from acute lactic acidosis in infancy with severe neurologic impairment in affected males to a slowly progressive neurodegenerative disorder in some males and more commonly females. Structural abnormalities, such as agenesis of the corpus callosum, are often present on neuroimaging (76). Epilepsy frequently occurs and includes infantile spasms and myoclonic seizures. EEG findings include multifocal slow spike–wave discharges (77). The ketogenic diet may benefit these patients.
Pyruvate Carboxylase Deficiency
Pyruvate carboxylase is a biotin-responsive enzyme that converts pyruvate to oxaloacetate in the citric acid cycle. Two predominant clinical presentations occur with pyruvate carboxylase deficiency. The neonatal type (type B) manifests with severe lactic acidemia and death in the first few months of life. The infantile and juvenile type (type A) begins in the first 6 months of life with episodes of lactic acidemia precipitated by an infection. Developmental delay, failure to thrive, hypotonia, and seizures, including infantile spasms with hypsarrhythmia, may be seen (78). A benign form (type C) also has been described with recurrent metabolic acidosis and normal neurologic development (79).
Seizures are related to the energy dysfunction that occurs secondary to Krebs cycle dysfunction. Treatment with the ketogenic diet or corticotropins may markedly exacerbate the disorder and should be avoided (80,81).
Leigh Syndrome
Leigh syndrome (subacute necrotizing encephalomyopathy) is both a clinical and radiologic phenotype and may be related to various metabolic defects, including syndromic and nonsyndromic mitochondrial disease, and PDH deficiency. Biochemical defects in both nuclear and mitochondrially encoded genes have been identified with this condition. It is genetically heterogeneous, and depending on the etiology, may be autosomal recessive or dominant, X-linked, or maternally inherited (82).
The clinical presentation is often acute to subacute, involving regression, cranial nerve and bulbar dysfunction, progressive hypotonia, lactic acidosis, and failure to thrive. The disease progresses with spasticity and central respiratory failure. Neuroimaging shows bilateral, fairly symmetric, basal ganglia, thalamic, and midbrain lesions that can fluctuate in severity. Varying degrees of white matter lesions may also be present along with cortical and cerebellar atrophy (83).
A variety of different seizures, including focal and generalized seizures, have been described (84). Infantile spasms and hypsarrhythmia may occur (85,86). In addition, there have been cases of epilepsia partialis continua (EPC) (87). EEG features do not appear to be distinctive enough to contribute to the clinical diagnosis of Leigh syndrome (88).
Disorders of Amino and Organic Acids Metabolism
Amino and organic acids predominantly form from the catabolism of proteins and carbohydrates. Any enzymatic defect in these metabolic pathways leads to an accumulation of potentially acidic compounds, and partial inhibition of the citric acid and urea cycles. Acidosis and hyperammonemia ensue leading to encephalopathy and, at times, seizures. These disorders, when most severe (a severe enzyme deficiency), typically present in the newborn period, especially after an infant is exposed to a protein or carbohydrate challenge in the diet. For some, this means after feeding in the first week of life, while for others it is after the introduction of solid foods. Regardless of the type of amino or organic acid disorder, the acute presentation is often the same. Milder enzyme deficiencies may present with a later sudden-onset epileptic encephalopathy (later infancy, childhood, or in the adult years) in the midst of a physiologic stressor (illness, surgery, fasting) that leads to accelerated catabolism. Thus, many of these metabolic disorders should be considered in a patient with an acute to subacute epileptic encephalopathy of later onset as well when an etiology for the problem remains unknown. With the advent and increased utilization of expanded NBS in states across this country, and internationally, many “classic” inborn errors of metabolism are now diagnosed and treated before they lead to neurologic symptoms. Conditions such as phenylketonuria (PKU), propionic or methylmalonic acidemia, and other relatively well-known amino or organic acid and fat metabolism disorders have become chronic conditions with improved neurologic outcomes due to early diagnosis and preventative care. However, there is no “standardized” NBS protocol, and the diseases screened for may vary from state to state and country to country. Thus, while many inborn errors of metabolism may now be excluded by the extended newborn screen, familiarity with the diseases screened for in one’s area is useful. As genetic knowledge of these conditions has evolved, we have moved from making an analyte-based diagnosis from blood and urine testing to confirmatory molecular genetic diagnostic studies. The treatment for all of these diseases is often very similar in the acute encephalopathy period—correct any metabolic derangements (acidosis/hyperammonemia), stop the introduction of the toxic substance by making the patient NPO, stop catabolism with dextrose-containing fluids, and prescribe any metabolic scavengers if needed. Below we discuss a few of the disorders where seizures are a prominent feature.
Phenylketonuria
One of the most frequent autosomal inborn errors of metabolism, occurring in 1 in 10,000 to 15,000 live births, PKU is caused by a deficiency in hepatic phenylalanine hydroxylase (89). As a consequence of the metabolic defect, toxic levels of the essential amino acid phenylalanine accumulate.
Currently, diagnosis is typically made with NBS, and in fact, this was the condition that led to the advent of NBS by Dr. Robert Guthrie in the 1960s. NBS is able to diagnose 100% of these patients. If the patient has not received NBS, routine amino acid analysis in plasma will identify elevated phenylalanine (90).
PKU occurs due to mutations in the PAH gene (12q23.2). Mutations in this gene account for more than 99% of cases (91).
If untreated, severe mental retardation, behavioral disturbances, psychosis, and acquired microcephaly can result. Seizures are present in 25% of affected children. The majority of children with PKU (80% to 95%) are also found to have abnormalities on the EEG. An age-related distribution of EEG findings and seizure types has been observed since a 1957 report by Low et al. (92). Infantile spasms and hypsarrhythmia predominate in the young infant. As children mature, tonic–clonic and myoclonic seizures become more frequent, and the EEG evolves to mild diffuse background slowing, focal sharp waves, and irregular generalized spike and slow waves (93,94). Donker et al. (95) showed proportionate increases in delta activity as levels rose during phenylalanine loading.
Primary treatment is a phenylalanine-free diet. With early detection and institution of this diet, the neurologic sequelae of hyperphenylalaninemia can be prevented or significantly minimized (96).
Maple Syrup Urine Disease
Maple syrup urine disease (MSUD) is an autosomal recessive condition due to a defect in the branched-chain α-keto acid dehydrogenase complex (BCKAD), and was first reported by Menkes et al. in 1954 (97).
The enzyme defect leads to accumulation of the branched-chain amino acids—valine, leucine, isoleucine—and their keto acids in body tissues and fluids. This disease is now tested for in most extended NBS panels and diagnosed prior to the onset of symptoms.
The BCKAD enzyme complex has four components, and three genes (BCKDHA/19q13.1–q13.2, BCKDHB/6p22–p21, and DBT/1p31) have been identified and mutations in these genes make up over 95% of cases (98).
Feeding difficulties, irritability, and lethargy are observed during the first few weeks of life. If left untreated, these signs may progress to stupor, apnea, opisthotonos, myoclonic jerks, and partial and generalized seizures. A characteristic odor can be detected in the urine and cerumen, but this may not be detectable until several weeks after birth. Milder variants of MSUD present with poor growth, irritability, or developmental delays later in infancy or childhood (1).
Laboratory testing reveals a metabolic acidosis and elevated blood and urine ketones. Hypoglycemia and hyperammonemia are rarely present in this disease. Ferric chloride testing of the urine causes a gray-green reaction, and the 2,4-dinitrophenylhydrazine test is positive. A marked elevation in branched-chain amino acids/branched-chain keto acids in the plasma, urine, and CSF amino acid profile is observed, and the presence of L-alloisoleucine is pathognomonic for this condition. Definitive testing can be performed by enzyme assay and molecular genetic studies (99).
The EEG shows diffuse slowing and a loss of reactivity to auditory stimuli. The “comb-like rhythm” characteristic of MSUD was initially reported by Trottier et al. in 1975, when bursts of a central mu-like rhythm were observed in four affected patients (100). Tharp described resolution of this pattern in an affected infant when dietary therapy was initiated (101). Korein et al. (100) observed a paroxysmal spike and spike–wave response to photic stimulation in 7 of 15 affected patients.
Pathologic studies reveal diffuse myelin loss and increased total brain lipid content. Cystic degeneration of the white matter associated with gliosis is observed. Disordered neuronal migration may occur with heterotopias and disrupted cortical lamination.
Acute treatment is aimed at counteracting the effects of hypoglycemia, acidosis, and ending catabolism. Dialysis or exchange transfusion rarely is necessary. Dietary therapy with protein restriction, thiamine supplementation, and elimination of branched-chain amino acids from the diet is the mainstay of treatment (99).
Histidinemia
Histidinemia or histidase deficiency is also associated with infantile spasms and myoclonic seizures. Other features include developmental delay and an exaggerated startle response. A diet low in histidine may be partially beneficial.
Isovaleric Acidemia
This condition is screened for by extended NBS. Symptoms develop during the neonatal period in half of the children with isovaleric aciduria (gene locus 15q14–q15). The presentation typically involves poor feeding, vomiting, dehydration, and a progressive encephalopathy manifested by lethargy, tremors, seizures, and coma. Depressed platelets and leukocytes may be seen, and the urine odor has been described as similar to that of “sweaty feet.” Cerebral edema is present, and seizures are most often partial motor or generalized tonic. The EEG shows dysmature features during sleep. Distinctive biochemical findings include metabolic acidosis, ketosis, lactic acidosis, and hyperammonemia. High urine concentrations of isovalerylglycine in urine organic acids and isovalerylcarnitine in acylcarnitine analysis are diagnostic. Genetic testing is clinically available (102).
Propionic Acidemia
This condition is screened for by extended NBS. The symptoms of propionic acidemia also appear during the neonatal period, with 20% of affected newborns having seizures as the first symptom. Characteristic features include vomiting, lethargy, ketosis, neutropenia, periodic thrombocytopenia, hypogammaglobulinemia, developmental retardation, and intolerance to protein. Patients may have very puffy cheeks and an exaggerated cupid bow upper lip. Mutations have been identified in both the α-subunit (13q32) and β-subunit (3q21–q22) of propionyl coenzyme A (CoA) carboxylase (103). Generalized seizures are typical, although partial seizures have also been reported. The EEG shows background disorganization, with marked frontotemporal and occipital slow-wave activity. In 40% of children, myoclonic seizures develop later in infancy, and older children may have atypical absence seizures. Biochemical findings include metabolic acidosis, ketosis, and elevation of branched-chain amino acids and propionic acid (104).
Methylmalonic Acidemia
This condition is screened for by extended NBS. Methylmalonic acidemia may be caused by deficiencies of the enzyme methylmalonyl-CoA mutase or adenosylcobalamin synthetic enzymes. Methylmalonic acidemia occurs in association with homocystinuria in the combined deficiency of methylmalonic-CoA mutase and MTHF: homocysteine methyltransferase (105,106). Forms responsive to vitamin B12 have been reported (107). Stomatitis, glossitis, developmental delay, failure to thrive, and seizures are the major features. Lesions of the globus pallidus on computed tomography or MRI are characteristic.
Diffuse tonic seizures and partial seizures with secondary generalization are the most frequent seizure types. Seizures may be characterized by eyelid clonus with simultaneous upward deviation of the eyes. In a review of 22 patients, Stigsby et al. (104) described abnormalities on the EEG in seven patients, consisting of multifocal spike discharges and depressed background activity in two, excessive generalized slowing in two, and mild background slowing with lack of sleep spindles in three. Two children were reported to have myoclonus and a hypsarrhythmic EEG pattern (108).
3-Methylglutaconic Aciduria
This condition is screened for by extended NBS. Severe developmental delay, progressive encephalopathy, and seizures are features of 3-methylglutaconic aciduria with normal 3-methylglutaconyl-CoA hydratase. This disorder results from a mutation on chromosome 9 in the gene encoding the enzyme 3-methylglutaconyl-CoA hydratase. Seizures occur in one-third of cases, and infantile spasms have been reported early in the course of the disorder. The typical organic acid abnormality includes marked elevations in 3-methylglutaconic acid and 3-methylglutaric acid in the urine (109).
3-Hydroxy-3-Methylglutaric Aciduria
This condition is screened for by extended NBS. Seizures are the presenting symptom in 10% of patients with 3-hydroxy-3-methylglutaric aciduria, a disorder caused by a deficiency in the enzyme that mediates the final step of leucine degradation and plays a pivotal role in hepatic ketone body production. The odor of the urine may resemble that of a cat. The chromosome location for this disorder is 1pter-p33 (110).
Glutaric Acidemia Type I
This condition is screened for by extended NBS. Glutaric acidemia type I is a more common autosomal recessive disorder of lysine metabolism that is caused by a deficiency in glutaryl-CoA dehydrogenase (19p13.2). Seizures are often the first clinical sign of metabolic decompensation after a febrile illness. Vigabatrin, L-carnitine, baclofen, and riboflavin supplementation have been suggested (111).
Urea Cycle Disorders
Urea cycle disorders (UCDs) occur due to partial or complete deficiencies of enzymes involved in the body’s attempt to remove waste nitrogen that forms from protein and carbohydrate catabolism. The incidence of these disorders is as high as 1:25,000, though later-onset diseases from partial defects are often underdiagnosed. All of these disorders are autosomal recessive except for ornithine transcarbamylase (OTC) deficiency which is X-linked dominant, but can present in both males and females. OTC is the most common of these disorders, accounting for over 50% of patients with UCDs (112).
The clinical manifestations of most of these disorders are similar and result, at least in part, from hyperammonemia. Typically, affected newborns present with poor feeding, emesis, hyperventilation, lethargy, or convulsions 1 to 5 days after birth. These signs lead to deepening coma, with decorticate and decerebrate posturing and progressive loss of brainstem function. Brain imaging and pathology reveal cerebral edema with pronounced astrocytic swelling (113). Later onset disease due to partial enzyme deficiencies can present with progressive spasticity of the lower extremity, episodic vomiting, or episodic fluctuating encephalopathy with or without seizures. Some individuals may be symptom free until in the midst of a physiologic stressor that leads to an acute metabolic decompensation (114).
The clinical diagnosis is confirmed by elevations in serum ammonia, absence of urine ketones, and respiratory alkalosis. In contrast, metabolic acidosis and ketosis frequently occur with disorders of organic acid or pyruvate metabolism. Characteristic findings in plasma amino and urine organic acids, along with measurements of urine orotic acid can help differentiate among the various enzymatic defects. Measurements of plasma citrulline and argininosuccinic acid may also be helpful. Definitive diagnosis is established via gene sequencing if the enzymatic defect is identified by screening biochemical tests in blood and urine. If the enzyme defect needs further defining or confirmation, biochemical analysis in skin fibroblasts or liver can be performed (115).
The EEG may show a low-voltage pattern, with diffuse slowing and multifocal epileptiform discharges (116). Two patients studied by Verma et al. (117) in 1984 demonstrated episodes of sustained monorhythmic theta activity. In patients with acute neonatal citrullinemia, a burst-suppression pattern has been described (118).
In the acute setting, hemodialysis, ammonia scavengers and protein restriction have been used to reduce serum ammonia and can be lifesaving. Protein restriction and medical therapy aimed at lowering serum ammonia are recommended in the long-term management of these children. Liver transplantation has been successful in correcting the enzyme deficiency, reducing ammonia levels, and reversing neurologic deficits in select patients (119,120).
Fatty Acid Oxidation Defects
The multienzyme, multistep process of fatty acid oxidation also occurs inside the mitochondria. Deficiencies of any of the enzymes involved, including carnitine palmitoyltransferase types I and II, may present in the newborn period (121). The infantile type of carnitine palmitoyltransferase II deficiency presents as severe attacks of hypoketotic hypoglycemia, sometimes associated with cardiac damage, culminating in sudden death. A deficiency in carnitine acylcarnitine translocase also may produce seizures, apnea, and bradycardia in the neonatal period. Seizures may infrequently occur in other defects of fatty acid oxidation, though they are not their most prevalent feature. When they occur, they are often due to associated hypoglycemia or hepatic dysfunction.
Peroxisomal Disorders
Peroxisomes play an important role in the body’s ability to break down very-long-chain-fatty acids (VLCFA) including pristanic acid via beta-oxidation and phytanic acid via alpha-oxidation. They are also involved in the biosynthesis of bile acids, plasmalogens, and pipecolic acid (122).
Disorders of the peroxisome have been divided into three categories: (i) disorders of peroxisomal biogenesis (Zellweger syndrome spectrum [ZSS]), (ii) disorders of a single peroxisomal enzyme (X-linked adrenoleukodystrophy [XALD], acyl-coenzyme A [acyl-CoA] oxidase deficiency); and (iii) disorders with deficiencies of multiple peroxisomal enzymes (rhizomelic chondrodysplasia punctata). The discussion that follows is limited to ZSS and acyl-CoA oxidase deficiency. XALD is discussed with the later-onset conditions (1).
Zellweger Syndrome Spectrum
The ZSS is the most common peroxisomal disorder in early infancy, with an estimated incidence of 1:50,000. This disorder was previously categorized as three distinct diseases (ZS, neonatal adrenoleukodystrophy, and infantile Refsum disease), but is now known to be a single condition with a varying spectrum of phenotypic severity. The ZSS phenotype is caused by mutations in any of several different PEX genes involved in peroxisome biogenesis, of which at least 12 have been identified. PEX1 mutations are the most common cause of ZSS (123).
Diagnosis is made via sending a peroxisomal enzyme or VLCFA panel that measures plasma levels of VLCFA, phytanic and pristanic acid, and plasmalogens. VLCFA alone can be the initial screening test, though one must keep in mind that the degree of elevation may vary and that false negatives can occur (124). Confirmatory molecular genetic testing is available as well.
Dysmorphic features may be noted shortly after birth. Within the first week to several months of life, the affected child develops encephalopathy, hypotonia, and hyporeflexia. Seizures occur in 80% of patients, including partial, generalized tonic–clonic (rare), and myoclonic seizures, and atypical flexor spasms. Multisystem abnormalities of the brain, kidneys, liver, skeletal system, and eyes may occur. Eye abnormalities include cataracts, glaucoma, corneal clouding, optic nerve hypoplasia, pigmentary retinal degeneration, and Brushfield spots. The presence of the latter, along with hypotonia and a dysmorphic appearance, may cause confusion in the diagnosis of Down syndrome versus ZSS. Findings on neuroimaging can include pachygyria or polymicrogyria localized to the opercular region and cerebellar heterotopias (125).
Patients with ZSS have partial motor seizures originating in the arms, legs, or face. The seizures do not culminate in generalized seizures and are easily controlled with medication. The interictal EEG of patients with ZS shows infrequent bilateral independent multifocal spikes, predominantly in the frontal motor cortex and surrounding regions. Less frequently, hypsarrhythmia is observed (126).
Presently, only symptomatic treatment is available for this condition.
Acyl-Coenzyme A Oxidase Deficiency
Acyl-CoA oxidase deficiency was initially described in two siblings by Poll-The et al. (127). Clinical features included hypotonia, pigmentary retinopathy, hearing loss, developmental delay, adrenocortical insufficiency, absence of dysmorphic features, and onset of seizures shortly after birth. A deficiency in acyl-CoA oxidase was identified, resulting from a deletion in its coding gene (17q25). In children with acyl-CoA oxidase deficiency, serum VLCFA levels are elevated, whereas pipecolic acid levels are normal. Cortical malformations are generally absent, and the interictal EEG may show continuous diffuse high-voltage theta activity (128).
Tay–Sachs and Sandhoff Disease
GM2 gangliosidosis is an autosomal recessive lysosomal disorder that invariably includes seizures as a prominent feature. The infantile forms of GM2 gangliosidosis include Tay–Sachs disease, caused by a deficiency in hexosaminidase A (Hex A), and Sandhoff disease, caused by a deficiency in Hex A and B. The enzymatic defect leads to intraneuronal accumulation of GM2 ganglioside. Tay–Sachs disease does not have any extraneural involvement, and the clinical presentation is that of a progressive encephalopathy (1).
The HEXA gene is localized to chromosome 15 (15q23–q24), and mutations are found more commonly in the Ashkenazi Jewish population of Eastern or Central European descent. The overall incidence of 1 in 112,000 live births in the general population increases to 1 in 3900 in this defined group (129).
Development appears to be normal until 4 to 6 months of age, when hypotonia and loss of motor skills are evident. Within the next several months to years, spasticity, blindness, and macrocephaly develop. The classic cherry-red spot is present in the ocular fundi of more than 90% of patients. Seizures become prominent, with frequent partial motor, complex partial, and atypical absence seizures that respond poorly to medication. Myoclonic jerks are frequent and are often triggered by an exaggerated startle response to noise (1).
The EEG is normal early in the course of disease. Gradually, background activity slows, with bursts of high-voltage delta activity and very fast central spikes. Diffuse spike and sharp-wave activity may be noted with acoustically induced myoclonic seizures. EEG amplitude declines as the disease progresses (130).
Enzymatic studies in leukocytes and skin fibroblasts reveal an isolated absence or deficiency in hexosaminidase A activity. Clinical genetic testing is available. Prenatal diagnosis and carrier detection for high-risk populations are also available (1).
Sandhoff disease is associated with a mutation of the β-subunit of hexosaminidase, (HEXB) located on chromosome 5 (5q13) (131). Unlike Tay–Sachs disease, there is no association with a particular ethnic group. Clinical presentation is similar to that of Tay–Sachs; however, distinguishing features in some patients include hepatosplenomegaly and skeletal involvement. Enzymatic testing demonstrates the diminished activity of hexosaminidase A and B. Detection of N-acetylglucosamine-containing oligosaccharides in the urine and foam cells in the bone marrow is also diagnostic. As with Tay–Sachs disease, no treatment is immediately available (1).
Krabbe Disease (Globoid Cell Leukodystrophy)
Globoid cell leukodystrophy (Krabbe disease) is another lysosomal disorder occurring in this age group and is due to a deficiency of galactocerebrosidase (GALC) enzyme activity. The GALC gene is located at chromosome 14q31 (132).
Depending on the severity of the enzyme deficiency, there are four phenotypic presentations of the disorder: (i) infantile onset, before the age of 6 months; (ii) late infantile onset, between ages 6 months and 3 years; (iii) juvenile onset, between ages 3 and 7 years; and (iv) adult onset, beginning after 7 years of age. The majority of cases begin within the first 3 to 6 months of life with irritability, poor feeding, emesis, and rigidity. Muscular spasms induced by stimulation are prominent. Blindness and optic atrophy ensue. Initially, increased tendon reflexes are present and then gradually diminish as breakdown of peripheral myelin occurs (133).
Partial or generalized clonic or tonic seizures, as well as infantile spasms, are seen, which may be difficult to distinguish from muscular spasms (134). In contrast to what is observed in many classic white matter diseases, seizures occur early in the course of Krabbe disease in 50% to 75% of infants with the disorder. EEG characteristics include a hypsarrhythmia-like pattern with irregular slow activity and multifocal discharges of lower amplitude than that typically seen with West syndrome. In a 1969 study of seven infants by Kliemann et al., six children had prominent β activity occurring independently in the posterior temporal regions and vertex that was superimposed over slower, high-amplitude waves. This activity was observed to be state dependent and to occur in long runs without any apparent clinical manifestations. In the terminal stages of the disease, little electrical activity is detected (135).
Diagnosis is made by measuring GALC enzyme activity in leukocytes or skin fibroblasts, followed by confirmatory gene sequencing. The disease is relentlessly progressive, with death by 1 to 2 years of age (133).
GM1 Gangliosidosis Types I and II
GM1 gangliosidosis occurs due to lysosomal β-galactosidase (GLB) deficiency, and mutations in the GLB1 gene localized to chromosome 3 (3p21.33). GLB deficiency leads to the accumulation of GM1 ganglioside and degradation products in nerve cells and other tissues. In the infantile form, the affected child is initially developing typically and then has a regression in development by 3 to 6 months of age, with rapid neurologic deterioration following. Seizures develop by 2 years of age. Clinical features may include coarse facial features, hepatomegaly, bone deformities (dysostosis multiplex), visual abnormalities, hypotonia, progressive microcephaly, and hematologic abnormalities. A macular cherry-red spot can be seen. Diagnosis is determined by findings of reduced GLB enzyme activity in leukocytes or skin fibroblasts, urine galactose-containing oligosaccharides in association with elevated keratan sulfate, vacuolization in blood lymphocytes or bone marrow, and distinctive findings on long bone and spine radiographs. Definitive testing is via molecular genetics (136).
The juvenile form of the condition typically has a slower neurologic progression. Cerebral manifestations with regression of developmental milestones and visual symptoms are typically present by 2 to 4 years of age (136).
EEG features of both forms include background slowing, with increasing, irregular slow activity as the disease progresses. In type II, a fluctuating 4- to 5-cycle temporal rhythmic discharge has been observed (137).
Progressive Encephalopathy with Edema, Hypsarrhythmia, and Optic Atrophy
Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome, described by Salonen et al. (138) in 1991, is characterized by infantile spasms, arrest of psychomotor development, hypotonia, hypsarrhythmia, edema, and optic atrophy. Characteristic features include epicanthal folds, midfacial hypoplasia, protruding ears, gingival hypertrophy, micrognathia, and tapering fingers. Edema develops over the limbs and face. The progressive decline seen with this disease suggests a metabolic defect, although no biochemical marker has been identified. Based on the pattern of inheritance associated with the disease, it is presumed to be an autosomal recessive disorder. Neuroimaging shows progressive brain atrophy and abnormal myelination. Hypoplasia of the corpus callosum has been reported. Seizures generally begin as infantile spasms with associated hypsarrhythmia on the EEG. Later, other seizure types may be seen, including tonic, tonic–clonic, and absence seizures. The EEG may evolve to a slow spike–wave pattern. Prognosis is poor in children with this disorder, with survival only into adolescence (139).
METABOLIC DISORDERS OF LATE INFANCY, CHILDHOOD, AND ADOLESCENCE
While many of the previously discussed disorders can have a later onset of symptoms, there are several disorders that typically begin outside of the newborn and early infancy period. A few of these disorders are discussed below.
Neuronal Ceroid Lipofuscinoses
The neuronal ceroid lipofuscinoses (NCLs) are a group of diseases that result in storage of lipopigments in the brain and other tissues. At least five clinical subtypes have been reported, as well as rare, atypical forms, and most are transmitted as autosomal recessive traits. The disorder can present at any age, from infancy through adulthood.
The condition occurs due to a genetic defect leading to impaired lysosomal function and intra- and extralysosomal storage. Thus far at least fourteen genes (CLN 1–14) have been identified, leading to the various phenotypes, though depending on the type of mutation, there is significant overlap between age of onset and symptoms (140).
Evaluation for this condition begins with quantification of palmitoyl-protein thioesterase (PPT1) and tripeptidyl-peptidase (TPP1) enzyme levels in leukocytes. Based on the enzyme deficiency found and age of onset, targeted gene sequencing is performed. If these levels are normal or the patient has an adult onset of symptoms, electron microscopy of skin for characteristic abnormalities and/or lymphocytes for vacuoles is recommended. This approach has evolved from the previous one of obtaining a skin biopsy as the diagnostic test in all patients. In rare instances, storage product identification in cells from a rectal suction or conjunctival biopsy is necessary. With advances in genetic knowledge, this type of testing is now infrequent. A next-generation sequencing panel of the various NCL genes is available (141,142).
Visual loss is a feature of almost all except the adult form of NCL either as the presenting symptom or occurring months to years after disease onset. The infantile form typically presents between 6 and 24 months of age with developmental regression, myoclonus, ataxia, and visual failure. Other features include incoordination of limb movements, acquired microcephaly, and optic atrophy. Seizures are prominent, including myoclonic jerks and astatic, atonic, or generalized seizures. EEG features aid in the diagnosis, with an early attenuation and progressive loss of the background. Neuroimaging may show progressive cerebral and cerebellar atrophy (143).
Epilepsy begins between the ages of 2 and 4 years in the late infantile form, followed by cognitive decline, ataxia, and eventually visual failure with optic atrophy. Early development is normal or mildly delayed. Multiple seizure types develop as well, with staring, generalized tonic–clonic, myoclonic, and atonic components. As the disease progresses, irregular myoclonus evoked by proprioceptive stimuli, voluntary movement, or emotional fluctuations become prominent. A characteristic EEG pattern of occipital spikes on low-frequency photic stimulation is observed. Giant visual evoked responses and somatosensory evoked potentials are seen as well. Juvenile-onset disease presents with visual loss between the ages of 4 and 10 years. Epilepsy begins a year to several years later, with multiple seizure types. The treatment of these conditions is symptomatic. Life span varies from several years to adulthood depending on the severity of the enzyme defect (143).
Metachromatic Leukodystrophy
Metachromatic leukodystrophy is the result of a deficiency of arylsulfatase A (ASA), leading to an accumulation of lipid sulfatide. Late infantile, juvenile, and adult-onset subtypes occur with about half of patients presenting between the ages of 1 and 2 years. Hypotonia, weakness, and unsteady gait suggestive of a neuropathy or myopathy are the most common presenting symptoms with the late-infantile form. These symptoms are followed by a progressive decline in mental and motor skills (144).
Partial seizures develop late in the clinical course in 25% of patients with the late-infantile form of metachromatic leukodystrophy and in 50% to 60% of patients with the juvenile-onset form (145,146). A progression from normal EEG features to diffuse slowing with epileptiform discharges correlates well with clinical decline (147). Bone marrow transplantation, especially if performed prior to the onset of neurologic symptoms, may be beneficial in some patients and may be accompanied by improvements in clinical neurophysiologic studies (148).
Diagnosis is initially made by quantifying ASA enzyme activity in leukocytes or cultured skin fibroblasts. Urine sulfatide activity can be measured. Molecular genetic testing of the ARSA gene (22q13.3–qter) is clinically available. Treatment is symptomatic (149).
Mucopolysaccharidoses
The mucopolysaccharidoses are a family of lysosomal storage disorders caused by a deficiency in several enzymes involved in the degradation of glycosaminoglycans. The various mucopolysaccharidoses share many clinical features, including a progressive course, multisystem involvement, organ enlargement, dysostosis multiplex, and abnormal facial features (150).
A subtype with a primarily neurologic phenotype is Sanfilippo syndrome (mucopolysaccharidosis type III), in which only heparan sulfate is excreted in the urine; four different forms have been described, each associated with a different enzymatic defect. The gene locus is 17q25.3 (151).
Generalized seizures develop in about 40% of patients with Sanfilippo syndrome, but these are often easily controlled with AEDs. Progressive dementia and severe behavioral disorders are other features. In a meticulous study of one patient, the EEG showed lack of normal sleep staging, absence of vertex waves and sleep spindles, and an unusual alteration of low-amplitude fast activity (12 to 15 Hz) with generalized slowing (152). Bone marrow transplantation was successful in several cases but not useful in others. Enzyme replacement therapy is available for some of these conditions (153).
Sialidosis Type I
Sialidosis type I, an autosomal recessive disorder of late childhood to adolescence, is characterized by progressive visual loss, polymyoclonus, and seizures. The myoclonus can be debilitating and is stimulated by voluntary movement, sensory stimulation, or excitement. Increased myoclonus with cigarette smoking and menstruation has been reported. As the disease progresses, cognitive decline, cerebellar ataxia, and blindness with optic atrophy occur. Dysmorphic features, bony abnormalities, and hepatosplenomegaly are absent. The EEG contains rhythmic spiking over the vertex, with a positive polarity overlying a low-voltage background (154). Neuroimaging shows diffuse cerebral and cerebellar atrophy. Diagnosis can be made by detection of an increase in sialic acid—containing oligosaccharides in the urine, vacuolated lymphocytes in the peripheral blood, and foamy histiocytes in bone marrow smears. Enzyme assays for deficiency of α-neuraminidase, the structural components of which are encoded on chromosome 10, offer definitive diagnosis. The gene defect has been localized to 6p21.3 (155).
Sialidosis Type II (Galactosialidosis)
Sialidosis type II, the juvenile form of this group of disorders, has features similar to those of sialidosis type I. Distinguishing characteristics are the less prominent myoclonic activity and the additional clinical features of coarse facies, corneal clouding, dysostosis multiplex, and hearing loss. Inheritance is autosomal recessive, and a higher incidence of this form of the disease is found in Japan. In the majority of cases, a partial deficiency of GLB can be seen in addition to neuraminidase deficiency (galactosialidosis), which may be the result of a defect in protective protein; the gene locus coding for this protein is 20q13.1. The EEG contains moderate-voltage generalized 4 to 6 per second paroxysms (156).
Gaucher Disease Type III
Three types of Gaucher diseases are known: type I, a chronic form with adult onset; type II, a rare form associated with infantile demise; and type III, a chronic form with neurologic involvement. These disorders result from a mutation in the gene encoding acid β-glucosidase (1q21), which leads to accumulation of glucosylceramide in the lysosomes of cells in the reticuloendothelial system (157). In the rare type III form, hepatosplenomegaly may be present from birth or early infancy, which may cause type III to be confused with the more common type I form of Gaucher disease. When neurologic symptoms develop in childhood to early adulthood, type III can be clearly distinguished from type I, in which cerebral features are absent. Frequent myoclonic jerks and tonic–clonic seizures ultimately develop. A supranuclear palsy of horizontal gaze is present in the majority of cases and is an important diagnostic sign. Generalized rigidity, progressive cognitive decline, and facial grimacing may be present. Paroxysmal EEG abnormalities may be seen prior to the onset of convulsions, with worsening as the disease progresses; diffuse polyspikes and spike–wave discharges are also seen. The most characteristic EEG findings are rhythmic trains of spike or sharp waves at 6 to 10 per second (158). The diagnosis can be made by the clinical findings in combination with Gaucher cells detected in the bone marrow. Another laboratory abnormality is an elevated serum acid phosphatase. Unlike type I disease, which is prevalent in the Ashkenazi Jewish population, type III is reported predominantly in Sweden. A multimodal approach is suggested with enzyme replacement therapy and deoxynojirimycin analogs aimed at blocking the synthesis of glucocerebroside to lessen the systemic manifestations. Therapeutic trials with bone marrow transplantation have shown some success in improving CNS manifestations of Gaucher disease type III (159).
Neuroaxonal Dystrophies
Axonal dystrophies include infantile neuroaxonal dystrophy, pantothenate kinase–associated neurodegeneration (formerly Hallervorden–Spatz disease), and Schindler disease. Pantothenate kinase-associated neurodegeneration is not discussed here, as seizures are not a prominent feature.
Infantile neuroaxonal dystrophy (Seitelberger disease) is an autosomal recessive disorder affecting both the central and the peripheral nervous systems. Characteristic pathologic features of axonal spheroids within the peripheral and central nervous systems are seen. Clinical features begin between 1 and 2 years of age with psychomotor regression, hypotonia, and development of a progressive motor sensory neuropathy. Seizures occur in one-third of patients, with onset of convulsions after 3 years of age. The EEG finding of high-amplitude fast activity (16 to 24 Hz), unaltered by eye opening or closure, is characteristic of all children with this disorder, regardless of the occurrence of seizures. During sleep, the fast activity may persist, and K complexes are typically absent (160). Seizure types described with infantile neuroaxonal dystrophy include myoclonic and tonic (161,162). A video–EEG case report by Wakai et al. (163) described tonic spasms and an electrographic correlate of a diffuse, one-second, high-voltage slow complex, followed by desynchronization suggestive of infantile spasms.
Schindler disease results from a deficiency in α-N-acetylgalactosaminidase (22q11). Affected patients appear normal at birth, but progressive neurologic decline becomes evident in the second year. Manifestations include spasticity, cerebellar signs, and extrapyramidal dysfunction. Generalized tonic–clonic seizures and myoclonic jerks are common. EEG abnormalities include diffuse and multifocal spikes and spike–wave complexes (164).
Mitochondrial Diseases in Older Infants and Children
An overview of mitochondrial disease in infants is outlined in an earlier portion of this chapter.
POLG1 Disease, Including Childhood-Onset Epilepsia Partialis Continua, and Alpers Disease
The mtDNA polymerase gamma (POLG) is a nuclear gene required for mtDNA replication. Over the recent past, mutations of this gene have been linked to a wide array of growing phenotypes. While many of these phenotypes do not have seizures as their primary or only feature, a phenotype with EPC as the initial and often only manifestation is known as is a form with progressive myoclonic epilepsy (165).
POLG mutations are also the principal cause of Alpers disease (166).
Alpers and Alpers–Huttenlocher diseases are characterized by a rapidly progressive encephalopathy with intractable seizures and diffuse neuronal degeneration. Seizures are often partial complex or myoclonic though they can evolve to include multiple types. The EEG may show rhythmic slowing, predominating either in the posterior or anterior derivations, sometimes admixed with periodic brush-like patterns (167). Varying amounts of liver disease is also present. Symptoms may begin at any age, and liver disease may not be present for years. Encephalopathy and liver disease can stabilize with partial resolution of symptoms. Disease onset after exposure to valproate and valproate-related worsening of existing symptoms is characteristic of this condition (168,169). There is recent debate about whether patients with encephalopathy and epilepsy of undetermined etiology need to receive POLG testing prior to initiating valproate therapy (170).
MERRF and MELAS
While we now understand that many patients with primary mitochondrial disease may not present syndromically or with maternally inherited disease, the initially described conditions designated by acronyms remain an important cause of mitochondrial disease and epilepsy. Two of these syndromic presentations of mitochondrial disease are described below.
Onset of MERRF occurs before 20 years of age, with ataxia and predominantly myoclonic seizures. Affected individuals may have short stature, neurosensory hearing loss, optic atrophy, myopathy, or encephalopathy. EEG findings may include background slowing, focal epileptiform discharges, and atypical spike or sharp and slow-wave discharges that have a variable association with the myoclonic jerks. Suppression of these discharges during sleep is characteristic. As with many of the progressive myoclonus epilepsies, giant somatosensory evoked potentials are observed. Lactic acidosis and the presence of ragged-red fibers on muscle biopsy are common features of the diagnosis. The inheritance pattern is compatible with maternal transmission. In the majority of cases, a point mutation at position 8344 of the mitochondrial gene for transfer ribonucleic acid (tRNA)-lysine has been identified (171). Other mtDNA point mutations are also known to lead to this phenotype.
Classically, mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) presents in childhood with the sudden onset of stroke-like episodes. Migraine-like headaches, progressive deafness, seizures, cognitive decline, and myopathic features may accompany these symptoms. EPC can be seen, and seizures often evolve into partial or generalized status epilepticus. Myoclonic seizures are prominent in individuals with MELAS. Strokes are often occipital in location, and seizures may present with visual auras and migraine. Lactic acid is elevated in the blood, and ragged-red fibers can be present on muscle biopsy. Four-point mutations are predominantly seen with MELAS. Three of these (3243, 3250, and 3271) affect the mtDNA gene of tRNA-leucine. The other mutation involves a coding region of complex I of the respiratory chain (172). Intravenous L-arginine has been used to prevent the morbidity of acute metabolic strokes in these patients, and its regular use has become more routine (173).
Dentatorubral–Pallidoluysian Atrophy
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare autosomal dominant disease due to a trinucleotide (CAG) expansion of the ATN1 gene on chromosome 12p (12p13.31). Clinical manifestations are dependent on the length of the unstable trinucleotide repeats and vary from a presentation of juvenile-onset progressive myoclonic epilepsy to an adult-onset syndrome with ataxia, dementia, and choreoathetosis. The juvenile form can also be variable in its presentation. In general, symptoms begin in infancy to early childhood with myoclonus, ataxia, dementia, opsoclonus, or seizures that can be generalized tonic–clonic, atypical absence, or atonic. Pathologic features are striking, with neuronal loss and gliosis in the dentatorubral and pallidoluysian structures (174).
The EEG characteristically shows bursts of slowing, irregular spike–wave discharges, and multifocal paroxysmal discharges. A photoparoxysmal response is seen, and myoclonic seizures can often be triggered by photic stimulation (175).
Congenital Disorders of Glycosylation
Congenital disorders of glycosylation (CDG) are multisystemic diseases characterized by a defect in the synthesis of N-linked glycoproteins and glycolipids. Over 50 disorders of glycosylation have now been identified leading to altered protein folding and stability (176).
CDG type Ia, phosphomannomutase-2 deficiency, is the best characterized of these syndromes (gene PMM2, 16p13.3–p13.2). The most common symptoms include infant-onset failure to thrive and hepatopathy. Developmental delays, cerebellar hypoplasia, ataxia, progressive neuropathy involving the legs, retinal degeneration, and skeletal deformities are also common. Subcutaneous tissue changes with an odd distribution of fat, retracted nipples, and odd facies, including almond-shaped eyes, have been described. Imaging studies reveal cerebellar hypoplasia. A unique pattern of coagulation changes is associated with the syndrome, including depression of factor XI, antithrombin III, protein C, and, to a lesser extent, protein S and heparin cofactor II. These changes may account for stroke-like episodes observed in affected children. Clinical neurophysiologic studies demonstrate interictal epileptiform discharges and giant somatosensory evoked potentials. Screening for this condition involves initially assessing transferrin glycosylation status via mass spectroscopy; more sensitive testing of a variety of glycosylated proteins via a mass spectroscopy N-glycan panel is also available (177). Mass spectroscopy testing is preferred in detecting less common disorders of glycosylation.
Disorders of Peroxisome Metabolism: X-linked Adrenoleukodystrophy
XALD is an X-linked genetic defect in one of the peroxisomal membrane transport proteins. The condition occurs due to mutations in the ABCD gene (Xq28) and leads to three varied phenotypes. The childhood onset form begins in early school age with attention deficit and cognitive regression.
Partial motor seizures, often with secondary generalization, and generalized tonic–clonic seizures can occur. Status epilepticus has been the initial presenting symptom, and EPC has also been reported. Diagnosis is made by quantifying VLCFA levels in plasma, followed by gene sequencing and confirmation (178). The EEG is characteristic, with high-voltage polymorphic delta activity and loss of faster frequencies over the posterior regions (179).
Adrenomyeloneuropathy and isolated Addison disease presentations account for the other phenotypes and are typically not associated with epilepsy.
Progressive Myoclonic Epilepsies
The progressive myoclonic epilepsies are a collection of disorders presenting with the triad of myoclonic seizures, tonic–clonic seizures, and neurologic dysfunction that often manifests as dementia and ataxia. Onset generally begins in childhood through adolescence, though they may begin later in life. If myoclonic features are not prominent, children with this syndrome may be erroneously diagnosed with Lennox–Gastaut syndrome. For this reason, a careful history to detect myoclonic features is important in children with intellectual deterioration and frequent seizures (180).
Previously discussed disorders that can lead to this phenotype include mitochondrial cytopathies (especially POLG mutations, MERRF, and MELAS), sialidoses, some of the other lysosomal diseases, DRPLA, and NCL. The two disorders below are listed separately since they are atypical storage diseases and are routinely tested for via direct molecular genetic testing.
Lafora Body Disease
Lafora body disease is an autosomal recessive disease that occurs due to mutations in either the EPM2A (6q24) or EPM2B (6p22.3) genes, encoding for the proteins laforin and malin (181). Laforin prevents the accumulation of phosphate on glycogen while malin is a ubiquitin ligase that regulates laforin. Defects in these genes lead to phosphate accumulation on glycogen and glycogen precipitation. Accumulation of this unbound glycogen leads to the formation of Lafora bodies and neuronal dysfunction (182).
Symptoms typically begin in the teen years, with focal, multiregional, or generalized myoclonus. The myoclonus is brought out by action, touch, light, and stress. Generalized tonic–clonic seizures may also occur. A prior childhood history of an isolated febrile or afebrile seizure may exist. Cognitive symptoms may lag by months or years and initially include visual hallucinations, personality changes, confusion, and ataxia. The visual hallucinations frequently represent occipital seizures (183).
Generalized bursts of spikes and polyspikes superimposed on a normal background may be seen initially on the EEG. The presence of spikes in the posterior quadrant is a distinguishing feature that suggests the diagnosis with the appropriate clinical scenario (184). As the disease progresses, the EEG becomes increasingly disorganized. A photoconvulsive response can be seen with photic stimulation.
On neuroimaging, cerebellar atrophy is occasionally observed. Intracytoplasmic inclusion bodies (Lafora bodies) are seen on electron microscopy of a skin, liver, or muscle biopsy. A negative biopsy does not exclude the diagnosis. Molecular genetic testing is the preferred route of diagnosis. There is no effective treatment for this disorder, and the average life span after onset is 2 to 10 years (183).
Unverricht–Lundborg Disease (Baltic Myoclonus)
This autosomal recessive progressive encephalopathy is characterized by relentless myoclonus and generalized seizures due to defective function of the cystatin B protein and mutations in the CSTB gene (21q22.3). Testing for a common dodecamer repeat (that leads to over 90% of cases) and gene sequencing is clinically available (185).
Onset is in childhood or adolescence with seizures that are predominantly myoclonic and frequently occur after awakening. Absence and atonic seizures are also observed. Myoclonus can become quite disabling, interfering with speech and swallowing, and is often provoked by voluntary movement and excitement. Cognition is generally retained, although a mild decline may be observed later in the disease course. A labile affect and depression are commonly seen. Cerebellar ataxia, tremors, hyporeflexia, wasting of the distal musculature, and signs of chronic denervation on electromyography may be seen as the disease progresses (186).
The EEG reveals progressive slowing, with generalized 3-to-5-per-second spike–wave-like bursts that are frontally predominant. Paroxysmal flicker responses and generalized spikes and polyspikes are seen with photic stimulation (187,188). Although this disorder occurs worldwide, it has an especially high incidence in Finland, Estonia, and areas of the Mediterranean.
Phenytoin and AEDs predominantly affecting sodium channels can worsen symptoms (189). Death occurs in the third to fourth decade of life.
Disorders of Amino Acid Metabolism: Homocystinuria
Disorders of transsulfuration include cystathionine β-synthase deficiency, the most frequent cause of homocystinuria; the gene locus is 21q22.3 (190). The condition is screened for by extended newborn testing in many states and thus treated presymptomatically. When untreated, mental retardation, behavioral disturbances, and seizures are manifestations of CNS involvement; ectopia lentis, osteoporosis, marfanoid habitus, and scoliosis are other common clinical findings (191). Some patients respond to pyridoxine therapy. Generalized seizures occur in about 20% of patients with pyridoxine-nonresponsive homocystinuria and in 16% of patients with the pyridoxine-responsive form. EEG features are relatively nonspecific, with slowing and focal interictal epileptiform discharges that may ameliorate with treatment (192). Thromboembolism, malar flushing, and livedo reticularis reflect vascular system involvement. Biochemical abnormalities include homocystinemia, methioninemia, decreased cystine concentration, and homocystinuria.
DIAGNOSTIC INVESTIGATION IN METABOLIC AND MITOCHONDRIAL DISORDERS
The diagnosis of genetically determined metabolic diseases can be complicated for many reasons. We suggest that a metabolic basis be considered for all unexplained epileptic conditions of infancy or childhood until proven otherwise. Routine interrogation of blood, urine, and CSF samples as discussed in this chapter will be informative in a significant subset of patients, and alternative treatment options may emerge. Before obtaining appropriate metabolic, biochemical, or tissue specimens, the physician should try to formulate a differential diagnosis. Age at onset, type of epilepsy, associated clinical findings, family history, ethnicity, and neurologic examination continue to be the most important considerations in initial diagnostic possibilities. Neurologists experienced in metabolic disorders can often narrow the list of possible disorders at the first clinical encounter. Therefore, a consultation with a metabolic specialist is useful before or after initial screening tests are performed.
The presence of macular cherry-red spots, abnormal appearance of the hair, or a peculiar distribution of fat over the posterior flanks or thighs immediately suggests a diagnosis of Tay–Sachs disease, Menkes disease, or CDG, respectively. Deceleration of head growth during infancy, with consequent acquired microcephaly, may imply Glut-1 transporter deficiency, another defect of energy metabolism, the infantile form of neuronal ceroid lipofuscinosis, or Rett syndrome, among other possibilities. Dislocated lenses and a seizure followed by a stroke are characteristic of homocystinuria and MOCOD. Seizures with stroke-like episodes also suggest CDG, mitochondrial disorders or OTC deficiency. Genetically determined metabolic diseases often have a saltatory historical pattern in contrast to neurodegenerative diseases, which are inexorably progressive.
Evaluation in the Absence of Overt Clinical Clues
In certain circumstances, the underlying problem will not be intuitively obvious and the patient’s disorder may masquerade as a form of cryptogenic epilepsy.
Certain screening tests can be used to help narrow the differential diagnosis. A complete blood cell count with differential and platelet count should be obtained in every case. Bone marrow depression occurs in the organic acidemias, and the peripheral smear may reveal important clues such as a macrocytic anemia or vacuolated lymphocytes. A complete serum chemistry profile will uncover acidosis, and electrolyte disturbances or specific organ dysfunction. A low blood urea nitrogen may suggest a defect involving the urea cycle. Calcium and magnesium concentrations should be determined in every case. A low uric acid concentration raises the possibility of MOCOD. Ammonia elevations point toward amino and organic acidopathies when mild, and urea cycle defects when marked. Quantitative measurement of plasma amino acids and urine organic acids provide diagnostic clues about disorders of amino and organic metabolism, mitochondrial disease, UCDs and disorders of vitamin metabolism.
When faced with refractory epilepsy without an etiology at any age, spinal fluid analysis is mandatory to exclude certain treatable causes of the epilepsy. An elevated CSF protein concentration is characteristic of demyelinating and inflammatory conditions, including certain mitochondrial disorders, metachromatic leukodystrophy and globoid cell leukodystrophy. A low CSF glucose concentration is consistent with hypoglycemia caused by a defect of gluconeogenesis or Glut-1 transporter defects. Lactate and pyruvate values are elevated in CSF disorders of cerebral energy metabolism, including PDH deficiency, pyruvate carboxylase deficiency, numerous disturbances of the respiratory chain, certain defects of neurotransmitter synthesis and Menkes disease. A low CSF lactate value may be seen in Glut-1 transporter defects. CSF amino acids can provide additional information in disorders of ammonia, amino acid, organic acid, and mitochondrial metabolism. Elevations in threonine can be seen in pyridoxal 5-phosphate-dependent seizures. Elevations in glycine may point toward glycine encephalopathy (NKHG). Abnormally low serine concentrations point to PHGD deficiency.
Spinal fluid neurotransmitter analysis should also be routine in these circumstances. This testing includes measuring the neurotransmitter amines, biopterin, neopterin, and 5-MTHF levels. A low CSF 5-MTHF concentration suggests a defect involving folate metabolism, whether due to systemic disease, another inborn error of metabolism or primary cerebral folate deficiency. Abnormal CSF dopamine, and serotonin metabolites, along with biopterin and neopterin help diagnose disorders of neurotransmitter synthesis. In addition, if a special peak of a yet unknown compound appears on high-performance liquid chromatograms (routinely performed as part of spinal fluid neurotransmitter testing), the finding is diagnostic of pyridoxine- and folinic acid–responsive epilepsy due to ALDH7A1 mutations.
More focused testing for specific disorders may also be needed. These include transferrin and glycan analysis via mass spectroscopy for disorders of N-glycosylation, urine S-sulfocysteine for sulfite oxidase or MOCOD, lysosomal enzyme analysis in leukocytes for various storage disorders or PPT1 or TPP1 levels in leukocytes for the NCLs. A peroxisomal panel in blood or at least VLCFA levels will help diagnose ZSS and XALD. In addition, urine oligosaccharides, sialic acid levels, and glycosaminoglycans help better elucidate the cause of certain storage disorders. Urine guanidinoacetate and creatine levels are sent for diagnosing disorders of creatine synthesis. Urine and spinal fluid pipecolic acid and α-AASA levels are elevated at times in pyridoxine-dependent epilepsy.
Tissue biopsy specimens also provide important information in establishing a diagnosis. Specimens of skin, peripheral nerve, and skeletal muscle may provide useful clues as well. These tissues can be sent for electron microscopy, histopathologic staining, and selective biochemical analysis. Only rarely are a liver or brain biopsy necessary. Rectal and conjunctival biopsies are infrequently performed.
At times, but less frequently, the EEG features may be sufficiently distinctive to suggest the diagnosis of a limited number of conditions (Table 31.2). In other disorders, the EEG features can help narrow the differential diagnosis. For example, a burst-suppression pattern is seen in patients with NKH, PKU, MSUD, and molybdenum cofactor/sulfite oxidase deficiency, in addition to other disorders. Some distinctive EEG features include a comb-like rhythm with 7- to 9-Hz central activity, which is seen in patients with MSUD and propionic acidemia; vertex-positive polyspikes, seen in sialidosis type I; bioccipital polymorphic delta activity, seen in XALD; and 16- to 24-Hz invariant activity, seen in those with infantile neuroaxonal dystrophy.
Table 31.2 Electroencephalographic Patterns and Their Associated Disorders
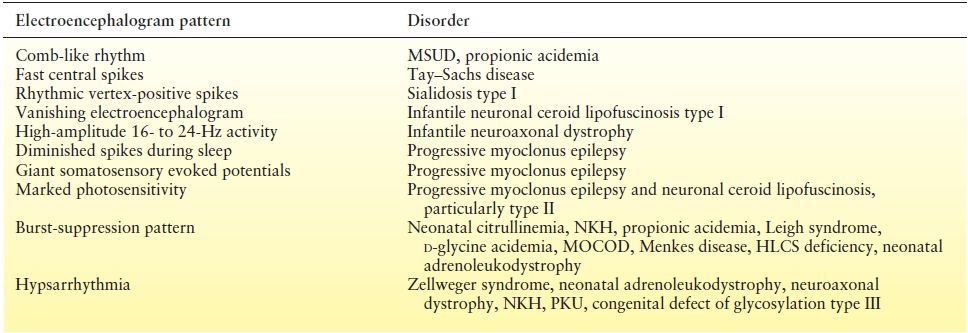
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


