Introduction
On January 26, 1841, Dr. W. J. West of Tunbridge, England, wrote a letter to the Lancet in which he described the condition of his son, then 1 year old.96 He wrote that the child was normal until 4 months of age, when he developed slight bobbings of his head forward. He initially regarded these head bobbings as a “trick”; however, West then observed that
these bobbings increased in frequency, and at length became so powerful as to cause a complete heaving of the head forward towards his knees, and then immediately relaxing into the upright position … these bowings and relaxings would be repeated alternately at intervals of a few seconds, and repeated from ten to twenty times at each attack, which attack would not continue more than two or three minutes; he sometimes has two, three or more attacks in the day; they come on whether sitting or lying; just before they come on he is all alive and in motion, making a strange noise, and then all of a sudden down goes his head and upwards his knees.
This is the first known written description of infantile spasms, and Dr. West could not have written a better description of the seizures. West went on to describe the decline in development of his son, stating that he “neither possesses the intellectual vivacity or the power of moving his limbs, of a child his age … he has no power of holding himself upright or using his limbs, and his head falls without support.”
Definitions
In recognition of Dr. West’s success at bringing this disorder to medical attention, the triad of infantile spasms, hypsarrhythmic electroencephalogram (EEG), and mental retardation is referred to as West syndrome. This chapter is devoted to the description of spasms as a particular type of epileptic seizure. In the literature, however, the term “infantile spasms” is used to indicate this form of seizure as well as a synonym for West syndrome. Consequently, in the literature it is impossible to distinguish the data referring only to this particular type of seizure from those referring also to the epileptic syndrome. The disorder is also referred to in the literature as massive spasms, salaam seizures, flexion spasms, jackknife seizures, massive myoclonic jerks, and infantile myoclonic seizures, in French as tics de salaam, and in the German literature as Blitz-Nick-Saalam Krampfe. Therefore, it was not clear from the beginning that these paroxysmal events were epileptic in nature.
The term “infantile spasms” is appropriate for a syndrome because it is characterized by the combination of a seizure type with a specific age of onset. The term “epileptic spasms” is more appropriate to refer to a seizure type. However, because “epileptic spasms” is not widely used, we will keep the term “infantile spasms” to refer to the seizure type occurring in infancy. Infantile spasms are considered to be generalized seizures and are said to resemble myoclonic seizures but with a tonic component.25 However, infantile spasm may begin with a focal onset.11,12,13 As noted by Carrazana et al.,13 “a wholly satisfactory categorization of infantile spasms is still elusive, reflecting our lack of understanding of the factors underlying this dramatic epileptic syndrome: namely how such a broad etiological range can be expressed in a common set of clinical and electrographic features.”
Following the latest classification of epilepsies and epileptic syndromes, infantile spasms as a syndrome have been conventionally classified into those in which there is no apparent preceding neurologic disorder or identified etiologic factor (idiopathic or presumably genetic in origin) and those in which a preexisting, presumptively responsible pathologic event or disorder is demonstrated (symptomatic).18 Some authors have further identified patients in the cryptogenic group in which, because of a genetic predisposition, an idiopathic etiology may be suspected.27,91 Because of the differences in prognosis, it is useful to classify the patients into those who were neurologically normal prior to their first infantile spasm (idiopathic–cryptogenic) and those who, for unknown reasons, were impaired prior to the first infantile spasms (symptomatic or probably symptomatic).
Epidemiology
Infantile spasms typically begin during the first 2 years of life, with a peak age of onset between 4 and 6 months of age. Approximately 90% of infantile spasms begin before 12 months of age, and it is rare for infantile spasms to begin during the first 2 weeks of life or after 18 months of age.44 However, cases with later onset, in the second year of life and up to 4 years of age, have been reported.6 The clinical and EEG features of infantile spasms beginning after the age of 12 months seem to be distinct, and it has been shown that they have features intermediary between West and Lennox-Gastaut syndromes. Although infantile spasms almost always begin in the early years of life, children resistant to treatment can continue to have the same spasms throughout infancy and adolescence and sometimes even during adult life.41
Clinical Features
Ictal
Although typically stereotyped in the same child, infantile spasms may vary considerably in clinical manifestations from one child to the next. Some seizures are characterized by brief head nods, whereas others consist of violent flexion of the
trunk, arms, and legs. As in Dr. West’s son, the spasms may evolve from slight head movements to more violent-appearing massive tonic events. The seizures are very brief, limited to rolling up of the eyes, and is particularly difficult to recognize in an infant who suffers from motor troubles such as spastic diplegia. Crying or irritability during a flurry of spasms is commonly observed, and thus spasms may be missed by casual observers, particularly when they express as cries that are believed to result from gastrointestinal reflux. The diagnosis often is dismissed by pediatricians and physicians who do not see the actual seizures.
trunk, arms, and legs. As in Dr. West’s son, the spasms may evolve from slight head movements to more violent-appearing massive tonic events. The seizures are very brief, limited to rolling up of the eyes, and is particularly difficult to recognize in an infant who suffers from motor troubles such as spastic diplegia. Crying or irritability during a flurry of spasms is commonly observed, and thus spasms may be missed by casual observers, particularly when they express as cries that are believed to result from gastrointestinal reflux. The diagnosis often is dismissed by pediatricians and physicians who do not see the actual seizures.
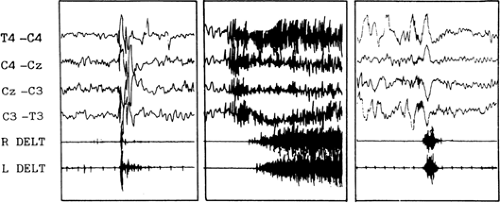 FIGURE 1. Polygraphic recordings of myoclonic, tonic, and spasm (left to right). Note that the duration of the spasms is intermediate between the myoclonic and tonic seizure. |
Although it resembles a myoclonic or tonic seizure, the spasm is a distinct type of seizure. As can be seen in FIGURE 1, a myoclonic jerk is a rapid, shock-like contraction of limited duration, whereas the tonic seizure is a prolonged muscle contraction of growing intensity. The true spasm consists of a characteristic muscular contraction that lasts from 1 to 2 seconds and reaches a peak more slowly than a myoclonic jerk but more rapidly than a tonic seizure. The electromyographic aspect is thus different from a myoclonic or atonic event.37
Kellaway et al.54 classified infantile spasms into three major groups: (a) flexor, (b) extensor, and (c) mixed flexor–extensor types. Flexor spasms consist of flexion of the neck, trunk, arms, and legs. Spasms of the muscles of the upper limbs result in either symmetric adduction of the arms in a self-hugging motion or adduction of the arms to either side of the head with the arms flexed at the elbow. Extensor spasms consist of a predominance of extensor muscle contractions producing abrupt extension of the neck and trunk with extensor abduction or adduction of the arms, legs, or both. Mixed flexor–extensor spasms include flexion of the neck, trunk, and arms and symmetric extension of the legs or flexion of the legs and extension of the arms with varying degrees of flexion of the neck and trunk. FIGURE 2 shows an example of symmetric mixed spasms in a cryptogenic patient.
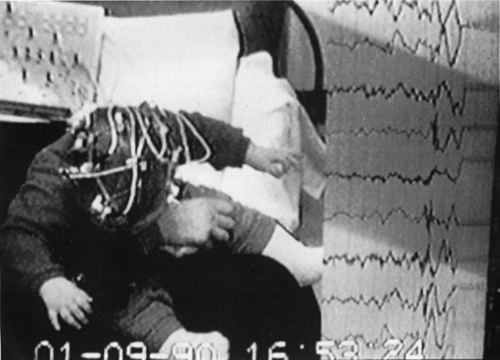 FIGURE 2. Clinical symmetric spasm in a cryptogenic patient. |
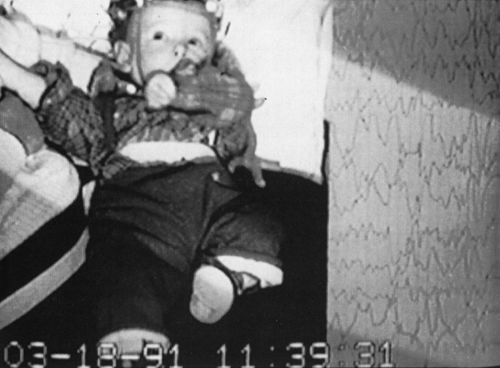 FIGURE 3. Clinical asymmetric spasm in a symptomatic patient. |
Asymmetric spasms can be seen when the contraction does not occur simultaneously on the two sides of the body (Fig. 3). This type of spasm is observed only in symptomatic infants with severe brain lesions or agenesis of the corpus callosum or both.37 Focal signs, such as eye or head deviation, may accompany symmetric and asymmetric spasms.92 Asymmetric spasms generally are isolated but can also follow a partial seizure, precede a partial seizure, or sometimes appear simultaneously with a partial or generalized seizure.
Momentary eye deviation or nystagmus may be a component of infantile spasms. Eye movements may precede the onset of the actual spasms by days or weeks or, in the patient with spasms, occur independent of the muscular contractions. At times, akinesia can occur independently without a preceding motor spasm. Sometimes, the clinical manifestations are very “subtle,” limited to yawing, gasping, facial grimacing, eye movements, or staring. Infantile spasms may also be associated with autonomic dysfunction characterized by pallor, flushing, sweating, pupillary dilation, lacrimation, and changes in respiratory and heart rate.60,62 Bisulli et al.9 studied muscular activation in symptomatic spasms by video and polygraphic recordings. They showed that the spasm is composed of a tonic and a phasic component; these two components are different events developing in variable order. In the same patient, spasms are clinically very similar, but the first activated muscle can vary in different spasms. The sequence and the latencies of muscle activation can also vary among different spasms. The absence of a rostrocaudal activation pattern is an element that characterizes the spasm as arising from aberrant cortical and subcortical connections and distinguishes the spasm from the cortical myoclonus.
Infantile spasms typically occur in clusters, and the intensity and frequency of the spasms in each cluster may increase in a crescendo fashion, peaking and then slowly decreasing in intensity. The number of seizures per cluster varies considerably, with some clusters having as many as 150 spasms. The number of clusters per day also varies, with some patients having as many as 60 clusters per day.
Postictal
Some children are irritable following a cluster of infantile spasms, whereas others demonstrate akinesia and impaired responsiveness for as long as 90 seconds after a flurry of seizures. Other children return to their baseline state immediately.
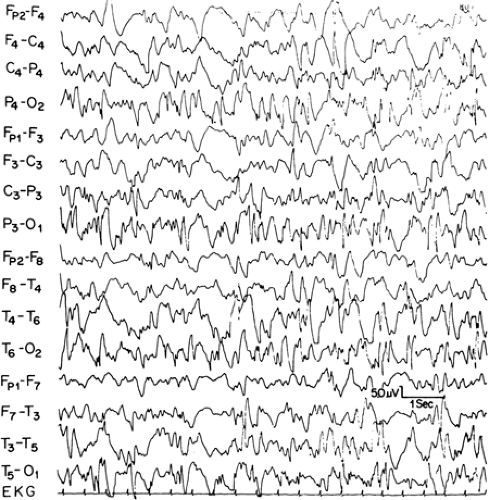 FIGURE 4. Example of a hypsarrhythmic electroencephalogram from a 4- month-old boy with tuberous sclerosis and infantile spasms. (From Carrazana EJ, Barlow JK, Holmes GL. Infantile spasms provoked by partial seizures. A case report. J Epilepsy. 1990;3:97–100, with permission.) |
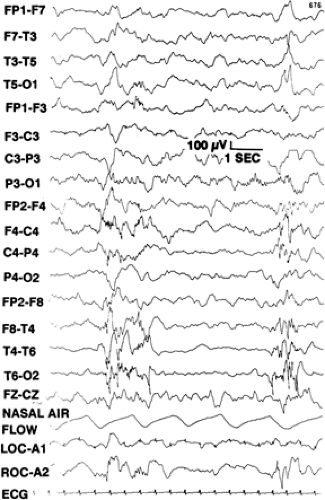 FIGURE 5. Example of an electroencephalogram (EEG) from a 14-week-old boy with infantile spasms and normal magnetic resonance imaging. Interictal EEG demonstrated burst suppression over the right hemisphere. The patient had partial seizures, which were characterized by a buildup of sharp theta waves over the right temporal-parietal-occipital region, which then evolved into a cluster of flexor spasms. The child underwent extensive resection of the parietal, occipital, and temporal lobes at age 7 months. Pathologic examination demonstrated cortical dysgenesis. (From Carrazana EJ, Barlow JK, Holmes GL. Infantile spasms provoked by partial seizures. A case report. J Epilepsy. 1990;3:97–100, with permission.) |
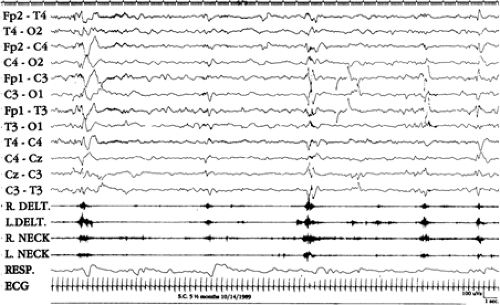 FIGURE 6. Example of a cluster of spasms in a cryptogenic patient. Note also the disappearance of hypsarrhythmia between one spasm and another. |
Interictal Development and Neurologic Function
Infantile spasms are frequently associated with developmental delay. In a review of the literature, Lacy and Penry60 reported that only 10% of patients were developmentally normal at the time of diagnosis of the infantile spasms. Patients with identifiable causes of infantile spasms, that is, those who are symptomatic, have a higher incidence of mental retardation than do cryptogenic cases.62
Neurologic abnormalities on physical examination are also commonly reported. Lacy and Penry60 reported that 70% of patients with infantile spasms have abnormal neurologic examinations. Children with identifiable etiologies for the spasms are
much more likely to have neurologic impairment than those in the idiopathic group.49,62 Lombroso62 found that 99% of patients with identifiable etiologies for the infantile spasms had abnormal physical examinations versus 20% of the patients with cryptogenic infantile spasms.
much more likely to have neurologic impairment than those in the idiopathic group.49,62 Lombroso62 found that 99% of patients with identifiable etiologies for the infantile spasms had abnormal physical examinations versus 20% of the patients with cryptogenic infantile spasms.
Electroencephalographic and Polygraphic Patterns
Interictal
Spasms are usually associated with markedly abnormal EEGs. The most commonly found EEG pattern is hypsarrhythmia (Fig. 4).38,73,94 This pattern consists of high-amplitude slow waves mixed with spikes and sharp waves whose amplitude and topography vary in an asynchronous manner between the two hemispheres.52 The background activity is completely disorganized and chaotic. During sleep, there are bursts of polyspike and slow waves. Somewhat surprising, in view of the marked background abnormalities, is the presence of sleep spindles in some patients. During rapid eye movement (REM) sleep there may be a marked diminution or complete disappearance of the hypsarrhythmic pattern.46,94 Infantile spasms are associated with a decrease of the total sleep time and a decrease in the percentage of REM sleep.45
Variations in hypsarrhythmia have been described. Hrachovy et al.46 reported a variety of forms of modified hypsarrhythmia, including patterns with interhemispheric synchrony, patterns with a consistent focus of abnormal discharge, recordings with episodes of attenuation, and hypsarrhythmia patterns consisting primarily of high-voltage slow activity with few sharp waves or spikes. FIGURE 5 shows an example of an EEG demonstrating a hypsarrhythmia pattern with burst-suppression over the right hemisphere.
Although a hypsarrhythmic or modified hypsarrhythmic pattern is the most common type of interictal abnormality seen in infantile spasms, this EEG pattern may not be present in some patients with infantile spasms.7,80 Although hypsarrhythmia was said to occur in other disorders as well,5,36,49 long video-EEG recordings show that patients with hypsarrhythmia usually exhibit spasms, although they may be difficult to identify behaviorally.
Ictal
As can be seen in FIGURE 2, the EEG equivalent of a myo-clonic jerk is a spike-wave or polyspike discharge, whereas tonic seizures consist of a recruiting rhythm or a polyspike discharge. Like the interictal pattern, the ictal EEG changes during infantile spasms are variable.37,54,56 For example, Kellaway et al.54 found 11 different types of ictal EEG patterns that accompanied the clinical seizures. In a careful study of the clinical and EEG-polygraphic features of 955 spasms in children with cryptogenic and symptomatic West syndrome, Fusco and Vigevano37 found that the most characteristic ictal EEG patterns of the spasms consisted of a positive wave over the vertex-central region, low-amplitude fast (14–16 Hz) activity, or a diffuse flattening called decremental activity.
Carefully studying the correlation between clinical manifestations and ictal EEG in 36 cases documented with video-EEG, Fusco and Vigevano37 established that the slow positive wave was present in all cases and always corresponded to the clinical manifestation of the spasm. The fast activity occurred alone or followed the slow wave. When it was alone, the main associated clinical feature was a motionless stare; this pattern sometimes appeared at the beginning or end of the cluster. Decremental activity was not always present. When it did appear it always followed the slow wave and clinical spasm. The decremental activity was never accompanied by clear behavioral events. Fast activity is of particular interest because it may have localizing value. Fast activity may express itself diffusely and symmetrically, asymmetrically, or sometimes with a focal predominance. In such cases the fast activity has a correlation with the cortical
lesion. Kobayashi et al.58 analyzed fast activity recorded by scalp EEG concurrent with spasms and established the correlation between asymmetry and the area of the lesion. According to Kobayashi and colleagues, this fast activity reflects the neocortical involvement in the genesis of the spasms. Consequently, spasms could be considered as a result of an interaction between cortical and subcortical systems.
lesion. Kobayashi et al.58 analyzed fast activity recorded by scalp EEG concurrent with spasms and established the correlation between asymmetry and the area of the lesion. According to Kobayashi and colleagues, this fast activity reflects the neocortical involvement in the genesis of the spasms. Consequently, spasms could be considered as a result of an interaction between cortical and subcortical systems.
The background EEG activity during a cluster of spasms may vary. In cryptogenic cases, hypsarrhythmia usually persists between one spasm and another; disappearance of hypsarrhythmia is observed in symptomatic cases and in infants with idiopathic spasms who have unfavorable outcomes.27,29 FIGURE 6 shows an example of a cluster of idiopathic spasms.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


