Evaluation of Intractable Epilepsy in Children
Epilepsy is the tendency to have more than one unprovoked seizure. The incidence of epilepsy is highest in the first years of life, and the majority of cases of childhood-onset epilepsy ultimately remit. Antiepileptic drugs (AEDs) remain the first line of treatment, with approximately 50% of patients achieving seizure freedom with their first medication trial and another 15% becoming seizure-free with a second medication.1 Unfortunately, in 15 to 30% of patients, epilepsy remains medically intractable, defined as a failure of adequate trials of two tolerated and appropriately chosen AEDs to achieve seizure freedom.2 Failure of therapy relies on the perception of the patient, parents, and physician that the medical treatment is not achieving an acceptable outcome. Infrequent seizures or a short duration of epilepsy does not exclude epilepsy from being intractable because both may still contribute to a significant medical risk. Even yearly seizures can exclude a patient from driving and adversely impact quality of life. Likewise, some patients present with frequent, disabling seizures for which intractability is rapidly determined, and surgical therapy may provide improvement in development. For those patients in whom intractable epilepsy develops, the evaluation can be extensive and is often carried out in specialized epilepsy centers well versed in the unique characteristics of pediatric epilepsy. The treatment strategies for such patients are complex and commonly include epilepsy surgery, neuromodulation, dietary therapy, and additional trials of medications used alone or in combination to achieve cure or palliation of symptoms.
71.1 Patient Selection
71.1.1 Pseudo-intractability
The process of labeling a child’s epilepsy as intractable may seem straightforward based on the operational definition of failure of two prior AED trials; however, one must first determine that the AEDs used were appropriate for the type of epilepsy. For example, the idiopathic generalized epilepsies typically respond very favorably to adequate treatment, and most treatment failures result from an inaccurate diagnosis of the epilepsy type or a poor choice of medication.3 Persistent uncontrolled seizures in a patient with juvenile myoclonic epilepsy who is on carbamazepine monotherapy should not be unexpected because this drug may worsen clinical seizures. Likewise, it is important to carefully delineate whether medication failure is secondary to adverse effects or whether failure has occurred after maximum dosing titrations, because tolerability of the treatment is a prerequisite for efficacy. In rare circumstances, the sedative effects of an AED can lower the seizure threshold and paradoxically increase seizure frequency. In a recent review of patients with pseudo-intractable epilepsy who ultimately became seizure-free following medication changes, therapeutic errors (i.e., inadequate dosing of medications) were implicated as the cause of intractability in 48% of cases, followed by inaccurate diagnosis of the seizure or syndromic type (47%) and medication noncompliance (14%).4
Equally important to ensuring that the treatments used were appropriately chosen and administered is demonstrating that the events being treated are epileptic in origin. Numerous behaviors incorrectly characterized as seizures can be encountered in children (see box “▶ Childhood Disorders That May Mimic Epilepsy”), and timely evaluation with video electroencephalography (EEG) can eliminate the need for an extensive evaluation of presumed intractable epilepsy.
Childhood Disorders That May Mimic Epilepsy
Behavioral disorders
Staring spells, inattention
Psychogenic nonepileptic seizures
Breath-holding spells
Self-stimulatory behaviors
Movement disorders
Focal dystonias
Nonepileptic myoclonus
Motor tics or stereotypies
Cardiogenic disorders
Vasovagal syncope
Arrhythmia (long QTc syndrome)
Other
Cataplexy
71.1.2 Assessment of Surgical Candidacy
Multiple historical features suggest a low likelihood of seizure remission. Most are recognizable early in the course of treatment, and the threshold for referring these children for further evaluation should be low (see box “▶ Historical Features That Predict Evolution to Intractable Epilepsy”). Childhood-onset epilepsy differs significantly from adult-onset cases, and several etiologies and syndromes are unique to the pediatric population (▶ Table 71.1). In many cases, the etiology may become clear only as the epilepsy evolves; thus, the history obtained by the clinician must include a detailed description of the seizures from the outset, such that idiopathic epilepsies of childhood, which ultimately remit, and those of symptomatic genetic origin (i.e., SCN1A mutations), which are unlikely to benefit from surgical therapy, are considered in the correct context. At the same time, there are several pediatric epilepsy syndromes and etiologies for which surgical therapy is especially efficacious and should be considered early in the course of treatment. Lastly, infants and young children differ in that catastrophic presentations of epilepsy may require more urgent surgical evaluation to prevent epileptic encephalopathy and loss of neurodevelopmental status.
Historical Features That Predict Evolution to Intractable Epilepsy
History of multiple seizure types49,50
History of infantile spasms50,51
Onset of seizures in infancy52,53
Remote symptomatic etiology51,52
Abnormal neurologic examination50,53
Frequent seizures (daily or weekly) before treatment50,52
Early recurrence of seizures (within 6 to 12 months of treatment)50
History of status epilepticus before diagnosis49–51
History of neonatal seizures49,51
Mental retardation49
Seizure clustering53
| Syndrome | Clinical findings | Recommended testing | |
| Dravet syndrome (severe myoclonic epilepsy of infancy) | Frequent or prolonged febrile seizures in infancy Myoclonic and hemiclonic seizures after the age of 1 year Developmental regression after onset | SCN1A gene testing | |
| Epilepsy and mental retardation limited to females | Frequent febrile seizures in infancy Frequent seizure clusters with tonic or generalized tonic–clonic seizures Myoclonic seizures less often than in Dravet syndrome Variable developmental delay | PCDH19 gene testing | |
| CDKL5 syndrome | Early onset of infantile spasms Evolution to multiple seizure types Encephalopathy | CDKL5 gene testing | |
| Autosomal-dominant nocturnal frontal lobe epilepsy | Early childhood focal seizures in non–rapid eye movement (REM) sleep Lifelong seizures with spontaneous remissions and relapses | CHRNA4, CHRNB2, CHRNA2 gene testing | |
| Etiologies of intractable epilepsy for which surgical therapy may be considered a treatment of choice54 | |||
| Low-grade cortical tumors Hippocampal sclerosis Focal cortical dysplasia |
71.2 Components of a Surgical Evaluation
Once intractable epilepsy has been diagnosed, the focus turns to determining an etiology, locating the source of the seizures, and evaluating the treatment options. A surgical evaluation is frequently undertaken, especially in patients with focal-onset seizures of symptomatic or cryptogenic etiology, for whom an excisional procedure may provide a cure. Even for patients unlikely to be cured, such as those with multifocal epilepsy, symptomatic or cryptogenic generalized epilepsy, or mixed seizure disorders, surgery may provide palliation, with efficacy equal or superior to that of alternative therapies.
The presurgical assessment should take place in a pediatric epilepsy center specializing in the evaluation of intractable epilepsy because the process is complex, and a team approach is required to develop successful treatment strategies. Every surgical evaluation must begin with a detailed description of each seizure type the patient has experienced because ictal semiology typically provides important localizing information (▶ Table 71.2). The ictal semiologies encountered in children are unique in that features may evolve throughout early childhood as the brain develops. In infancy, complex behavioral changes and stereotyped motor manifestations are less frequently encountered, likely because of immature neuronal networks.5 Typical lateralizing features of adult epilepsy (i.e., head version) are of less value in infants; the automatisms of infants are more often characterized by chewing or sucking, unlike the more stereotyped behavioral manifestations observed in older children and adults.6–8
| Semiologic pattern | Commonly associated lateralization/localization |
| Auras | |
| Psychic (emotion, déjà vu) | Temporal lobe, limbic system |
| Nausea | Insular cortex, frontal operculum, mesial temporal lobe |
| Olfactory, gustatory sensations | Limbic system, olfactory bulb, insular cortex |
| Ictal features | |
| Head version | Contralateral hemisphere, motor area anterior to precentral gyrus |
| Eye version | Contralateral hemisphere, frontal eye field |
| Unilateral clonus | Contralateral hemisphere, primary motor cortex |
| Homonymous positive visual phenomena | Contralateral primary visual cortex |
| Asymmetric tonic posturing (flexion of one upper extremity, extension of the other) | Supplementary motor cortex contralateral to extended upper extremity |
| Unilateral eye blinking | Ipsilateral hemisphere |
| Unilateral automatism | Ipsilateral mesial temporal lobe or contralateral temporal neocortex |
| Unilateral somatosensory paresthesias | Contralateral somatosensory cortex or supplementary sensory motor area |
| Postictal features | |
| Receptive aphasia/dysphasia | Temporal lobe, dominant hemisphere (Wernicke area) |
| Expressive aphasia/dysphasia | Inferior frontal gyrus, dominant hemisphere (Broca area) |
| Hemiparesis/hemiplegia | Contralateral hemisphere, motor cortex |
| Source: Foldvary-Schaefer N, Unnwongse K. Localizing and lateralizing features of auras and seizures. Epilepsy Behav 2011;20(2):160–16655 and Loddenkemper T, Kotagal P. Lateralizing signs during seizures in focal epilepsy. Epilepsy Behav 2005;7(1):1–17.56 | |
Intrinsic cerebral networks that inhibit seizure propagation develop over the first decade of life, at which time the typical lateralizing patterns of adult epilepsy begin to emerge. When patients are being evaluated for palliative surgical procedures, it is important to understand the frequency for each seizure type and the impact of the seizure type on quality of life. For example, while atypical absence seizures may be the most frequent seizure type in patients with Lennox-Gastaut syndrome, it is atonic seizures that lead to significant craniofacial injury and for which surgical treatment is often efficacious.
A detailed EEG evaluation, preferably with video, provides the foundation for most surgical evaluations. Children with a well-localized unilateral ictal onset more often attain seizure freedom, especially in cases with concordant interictal discharges and a recognized cerebral lesion on anatomical or functional imaging.9 Although patients may present with apparently generalized or multifocal EEG patterns, correlation with detailed anatomical magnetic resonance (MR) imaging and multiple functional imaging modalities may reveal a localized onset.10 Several anatomical and functional studies are often required in an epilepsy evaluation, each with inherent strengths and weaknesses with regard to the spatial and temporal localization of ictal onset and functional cortex (▶ Table 71.3).
| Modality | Spatial resolution | Temporal resolution | Contribution |
| Scalp electroencephalography (EEG) | Poor | Very good | Localization of seizure onset and interictal regions of irritability/dysfunction |
| Magnetic resonance (MR) imaging | Very good | Poor | Anatomical definition of epileptogenic lesions |
| Positron emission tomography (PET) | Good | Good | Localization of focal regions of hypometabolism corresponding to epileptogenic cortex |
| Single photon emission computed tomography (SPECT) | Good | Good | Localization of focal hyperperfusion corresponding to ictal onset |
| Magnetoencephalography (MEG) | Good | Very good | Localization of spike clusters with dipole maps, mapping of eloquent cortex |
| Functional magnetic resonance imaging (fMRI) | Good | Good | Mapping of eloquent cortex |
| Diffusion tensor imaging (DTI) | Good | Good | Mapping of white-matter tracts |
Anatomical high-resolution MR imaging with thin-slice volumetric T1-weighted gradient-recalled echo, in addition to axial and coronal T2, fluid-attenuated inversion recovery (FLAIR), and high-resolution T2 coronal imaging of the hippocampus provides excellent spatial localization of epileptogenic cortex and a basis for co-registering other imaging modalities. The most commonly encountered etiology of intractable focal epilepsy in children is focal cortical dysplasia, reported in more than 50% of cases in pediatric surgical series.11,12 Features of focal cortical dysplasia on MR imaging include focal cortical thickening, blurring of the gray–white junction, and increased gray matter signal on T2 or FLAIR imaging. The transmantle sign, characterized by a hyperintense white matter streak extending radially from the cortex to the lateral ventricle, may be encountered in higher-grade focal cortical dysplasia13–15 (▶ Fig. 71.1). Some of these features are more clearly delineated with postprocessing techniques, such as voxel-based morphometry. The addition of 3-tesla MR imaging improves anatomical definition and will identify lesions in 65% of patients with previously reported negative findings on MR imaging.16 Difficulties may arise, however, when the substrate for epileptogenesis has a very subtle or no abnormality on MR imaging, as may occur in cryptogenic or nonlesional epilepsy. It is increasingly recognized that successful anatomical localization depends on multimodal analysis with the co-registration of other modalities, such as positron emission tomography (PET) and single photon emission computed tomography (SPECT).13 Co-registration techniques may even identify epileptogenic zones beyond those identified with MR imaging, as has been demonstrated with PET and magnetoencephalography (MEG).17,18
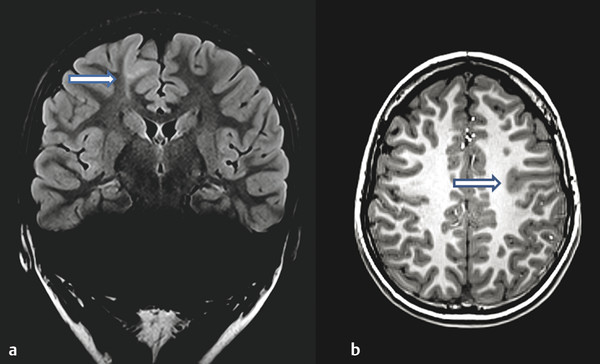
Fig. 71.1 Comparison of anatomical imaging characteristics of focal cortical dysplasia. (a) Example of transmantle sign (arrow) often encountered in type 2B cortical dysplasia (1.5 T [tesla], TR [repetition time] 9000, TE [echo time] 94, 3-mm slice thickness). (b) Example of cortical thickening and blurring of gray–white interface (arrow) seen in cortical dysplasia (3 T, TR 2100, TE 2.93, 1-mm slice thickness).









