While entire volumes have been devoted to the subject of model development in epilepsy (4,5), we will review the literature involving only a subset of the issues pertinent to a text on clinical epilepsy. Following an outline of techniques and the advantages and disadvantages of in vitro versus in vivo models, we will focus on the methods for invoking status epilepticus (SE) (a “prolonged” single seizure) via chemoconvulsants; single, repetitive, or prolonged seizures via hypoxia, temperature, kindling, or chemoconvulsants; and seizures induced by trauma or genetic alterations.
The process by which the initial insult (seizure, SE, or other) may lead to SRS (epilepsy) has been the subject of intense study, and multiple reviews have been put forth (6,7). Consensus regarding the relationship (cause or effect?) of hippocampal sclerosis and network reorganization to this process has not been forthcoming. Overall, the field has significantly shifted from a descriptive to a mechanistic focus involving key receptors, enzymes, and genetic regulation.
Seizures can be defined as paroxysms of abnormal, rhythmic, and synchronized discharges in the brain. Communication in the nervous system is a combination of electrical and chemical signaling with a balance between excitation and inhibition in each, primarily mediated between neurons. Glia modulate both types of communication primarily on a local basis, but frequently with distant consequences.
The resulting cascade (Fig. 3.1), beginning with receptor activation, followed by alterations in membrane polarization, potentially loops around, to result in alterations of the properties of the initial trigger of receptor activation. Consideration of this simplistic mechanism is important. Such a loop likely underlies normal plasticity associated with processes like learning and memory but perhaps becomes unstable with seizures and epileptogenesis, leading to aberrant plasticity that could result in both additional seizures and cognitive dysfunction.
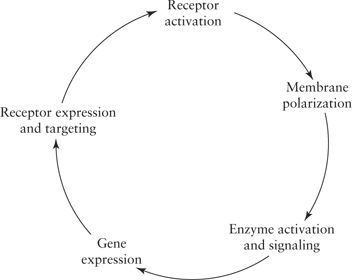
Figure 3.1. Proposed cascade of events following a seizure leading to potential adverse sequelae (medically resistant status epilepticus, epileptogenesis, learning impairment, etc.).
Review of Techniques
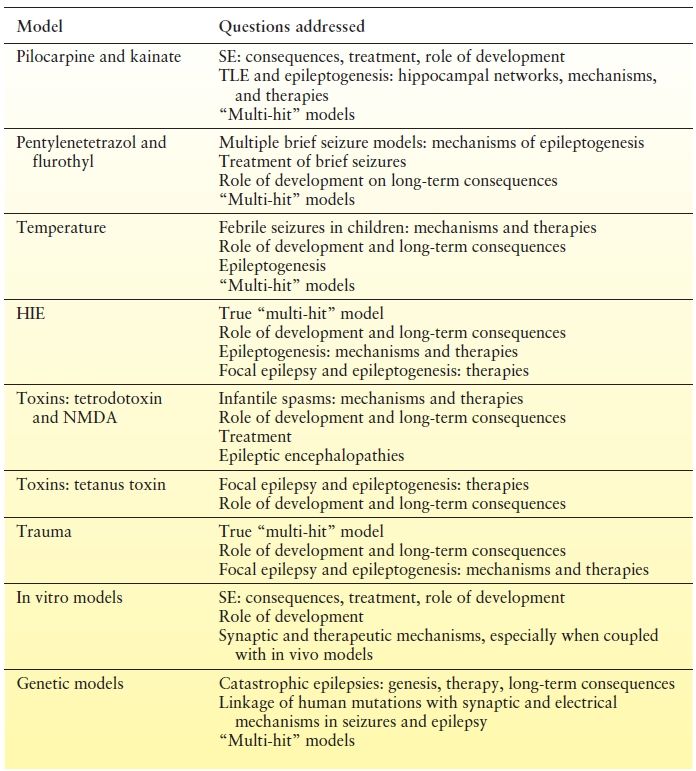
Experimental models can be divided into whole-animal (in vivo) versus in vitro studies (Table 3.2). Whole-animal models of acquired epilepsies typically involve single or multiple treatments to the animal that produce some form of injury or stimulation that results in later development of spontaneous seizures. Examples of these induced injuries include SE (chemoconvulsant and electrical), kindling, hypoxia, and head trauma. In genetic models, a spontaneous or induced genetic mutation or deletion results in seizures that happen spontaneously.
Table 3.2 Animal Model Summary
Seizure “activity” must be carefully defined for several reasons. First, the definition of a seizure is often extremely variable, as in the clinical literature. Second, consciousness, routinely used as a modifier in describing clinical seizures, is arbitrarily defined in most animals used. Typically, rhythmic, stereotyped, altered behavior is observed and characterized as a seizure. As in the clinical literature, EEG has become the gold standard for correlating altered behavior with seizures, but its use is limited due to the time- and labor-intensive placement of electrodes, limitations of electrode stability over time, and the fact that electrographic seizures emanating from deeper structures can be missed when recording from the cortical surface.
In Vitro Versus In Vivo Models
In vitro models involve removal and subsequent manipulations of whole-brain structures, slices of brain structures, or isolation and culture of separated brain cells (neurons and glia). These studies allow detailed manipulations and measurements but are limited in multiple, key ways. While it is tempting to designate repetitive electrical discharges as a seizure in these models, seizures in the whole animal are associated with a change in behavior or sensation, which cannot be appreciated in these in vitro models. Therefore, to avoid confusion, the repetitive electrical discharges must be referred to as “seizure-like” events or an ictus. It is important to note that one researcher’s abnormal ictal-induced phenomena may also be interpreted as another researcher’s normal activity-dependent changes. In addition, certain seizures, and their sequelae, may involve the interplay of multiple brain structures and are thus difficult if not impossible to recreate in in vitro models. Finally, key processes such as development and epileptogenesis, which occur over a prolonged period of time, cannot be fully studied in in vitro models as they are limited by the length of time the in vitro preparation is viable (hours to weeks).
There are dozens of in vivo and in vitro models of seizures and epilepsy, and, as already mentioned, there is little consensus about which if any are the “optimal model.” In reality, each model has its strengths and limitations, and the relative benefits depend on the specific question being asked. Below, we focus on the models that are in common use or emerging.
In Vivo Models
Pilocarpine and Kainate Models
The pilocarpine model and lithium–pilocarpine model [reviewed in (8)] involve the systemic administration of the muscarinic acetylcholine receptor agonist (pilocarpine) to induce a prolonged electrographic and behavioral seizure that requires cessation by benzodiazepines or barbiturates, typically after 1 to 2 hours, in order to prevent animal mortality. Clearly, from a clinical standpoint, muscarinic acetylcholine receptor agonism is very rarely the cause of SE in humans, with the exception of poisonings and chemical weapon exposures. Nevertheless, it is widely used because it results in severe SE, and animals eventually develop an epileptic phenotype with features very similar to those of human temporal lobe epilepsy (TLE) resulting in its widespread use for studying both of these conditions.
Kainate, a glutamate analogue that is not metabolized and can be injected either systemically or directly into the brain, results in seizures lasting several hours (9,10). Clinically, kainate originates as a shellfish poison, and human toxicity during outbreaks results in seizures and, in severe cases, hippocampal sclerosis (11). While this clinical situation is extremely rare, conditions involving glutamate overload that are known to be associated with seizures such as stroke, hypoxia (12,13), or infection may be mimicked to some degree by kainate administration. Similar to the pilocarpine model, kainate can induce an SE, and adult animals that survive kainate-induced SE may eventually develop an epileptic phenotype with features very similar to human TLE. In younger animals, kainate primarily activates the hippocampus, while in older animals, its effects are widespread (14).
Brief Seizure Models
Pentylenetetrazol and flurothyl are GABA-ergic antagonists that are administered systemically or inhaled, respectively (15). They both induce relatively short seizures, with flurothyl-induced seizures being very brief and limited nearly to the length of exposure to the vapors. As a result, both agents are used to mimic conditions involving single or multiple brief, generalized seizures (16). The major limitations of these models are that the mechanism of seizure induction does not clearly parallel any human condition, and the animals never develop spontaneous seizures. Both agents are thought to act on all susceptible brain regions, including cortex and hippocampus (15).
Electrical kindling, whereby electrodes are implanted in order to stimulate select brain regions, can also be used to study how repeated, brief seizure-like activity can influence outcomes. Depending on the stimulation protocol, kindling can eventually lead to induced behavioral seizures. This model, however, is limited by the technicalities of long-term implantation in rodents and the fact that most kindling paradigms do not result in development of spontaneous seizures.
Clinical Models: Fever and Hypoxia/Ischemia
In models where seizures are induced in the setting of increased temperature (fever), hypoxia, and/or ischemia, the ability of these models to generalize to human pathologies is clearly evident. Hypoxia models can involve placing animals in an environment of reduced oxygen content until seizures are observed (17,18). Other methods involve single or multiple cerebral vessel occlusions, often in combination with exposure to an environment with reduced oxygen content. Methods involving vessel occlusion are often time-intensive. These methods are then limited by the elements of hypoxia and ischemia, as these may independently influence outcomes (19).
Temperature-induced seizures in developing animals (20,21) involve slowly heating the animal, typically with warmed air, until seizures are initiated. This model is gaining popularity as a model of febrile seizures but may be limited by the fact it is really a model of externally imposed hyperthermia rather than endogenous fever as occurs in the human condition. These models are particularly attractive as they may be used as preclinical models to evaluate therapies.
Toxin Models
Several models involve the direct infusion of toxins, compounds, or even genetic material into specific regions such as the hippocampus. These are each meant to model focal seizures or epileptogenesis, though the result can have distant effects. These include the tetanus toxin model [reviewed in (22)] and more recently the tetrodotoxin model (23), thought to be a model of epileptic spasms or West syndrome. The knockdown of AMPA receptor GluA2 subunit by injection of antisense probes results in acute seizures (24). Following withdrawal of direct injection of glutamate receptor antagonists, spontaneous seizures are provoked in immature animals, while systemic injection does not cause this to happen (25).
Trauma Models
Experimental models of trauma utilizing either direct impact methods (26) or surgical undercuts (27) are gaining popularity as models for studying the development of posttraumatic epileptogenesis and epilepsy. As head trauma is a common cause of acquired epilepsy in humans, these models seem very generalizable to human pathology. As a result, these models have been used to study the efficacy of antiepileptogenic compounds as well as the mechanisms underlying posttraumatic epileptogenesis.
In Vitro Models
In vitro methods involving brain slices or cultures use a variety of methods to induce seizure-like electrical events. These can involve perfusion of compounds that typically enhance or favor membrane excitability alone or in combination with electrical stimulation, akin to kindling. The resulting spontaneous neuronal-mediated discharges can then be recorded from groups of neurons or from individual neurons typically using electrophysiologic techniques. Imaging techniques using fluorescent dyes that are able to indicate changes in membrane voltage or secondary changes due to accumulations of specific ions, such as calcium, often complement electrophysiologic measurements as they are able to simultaneously record from populations of neurons that may be somewhat distant from each other. The pattern of these discharges is then interpreted in isolation, in groups or bursts, or when the bursts cluster together as an ictus. The transitions between these types of discharges are interpreted as indicative of ictal genesis and are thought to generalize to seizure genesis. When the ictus is prolonged, this event generalizes to SE. When the ability to generate an ictus becomes more facile, this situation is thought to generalize to epileptogenesis. Determining how excitation spreads through a slice of brain tissue is generalized to how it may spread in the intact preparation. Thus, application of anticonvulsants to an in vitro preparation has been used to determine their efficacy and precise mechanism(s) of action. In order to circumvent the issues of truly generalizable seizures, SE, or epileptogenesis in vitro, brain slices are often prepared at various time points after these phenomena have developed in vivo. Findings from hippocampal brain slices prepared from animals after experiencing an induced or spontaneous seizure in vivo allow examination of how overall synaptic transmission, plasticity, and seizure thresholds have become altered by these processes.
Preclinical Models and New Therapy Development: Neonatal Seizures
As an example of the use of preclinical models, the parameters for establishing an antiseizure medication as first-line treatment for neonatal seizures have recently been considered (28,29). These include utilizing relevant rodent model(s) of neonatal seizures, using EEG confirmation of seizures and drug efficacy, dose–response evaluation of efficacy, and acute and long-term aspects, including later epilepsy and behavioral consequences. Determining the relevant model is nontrivial; as noted, no model is ever perfect. These issues include face validity (similar endophenotype in humans and rodents, i.e., does the EEG and clinical picture look similar?), construct validity (similar biologic dysfunction in humans and rodents, i.e., are the triggering mechanisms similar?), and predictive ability (similar biologic response to treatments by humans and rodents, i.e., are acute and chronic aspects similar?).
We reviewed 32 studies that involved 62 separate treatment trials from 1992 to present in postnatal day (P)20 or younger rodents (Table 3.3). Most studies use pretreatment with an antiseizure medication resulting in a lack of construct validity since prophylaxis is not used clinically. Few studies validated their findings with EEG, which is important since some drugs may be proconvulsant (NMDA receptor antagonists) and because antiseizure medications are known to cause “electroclinical dissociation” in neonates, resulting in attenuation of behavioral manifestations without reduction in electrographic seizure activity. Unfortunately, no studies validated with EEG and dose response, because lower doses were sometimes found to be more effective (clinically) and earlier treatment was sometimes more effective (by EEG). Importantly, phenobarbital, the most widely used therapy for neonatal seizures, has not been evaluated in the hypoxia model, the neonatal seizure model with likely the greatest construct validity. In the carotid artery occlusion model, phenobarbital had clinical efficacy when used as pretreatment. The efficacy of phenobarbital posttreatment has only been evaluated with EEG in the kainate model. The EEG efficacy of antidiuretic bumetanide, which is posited to overcome the depolarizing effect of GABA in the developing brain, has only been evaluated in the kainate model as pretreatment. Topiramate, levetiracetam, talampanel, and AMPA receptor antagonists have been evaluated for clinical efficacy (EEG data for levetiracetam) in the hypoxia model only when given as pretreatment.
Table 3.3 Evaluation of Preclinical Data for Anticonvulsant Efficacy for Neonatal Seizures
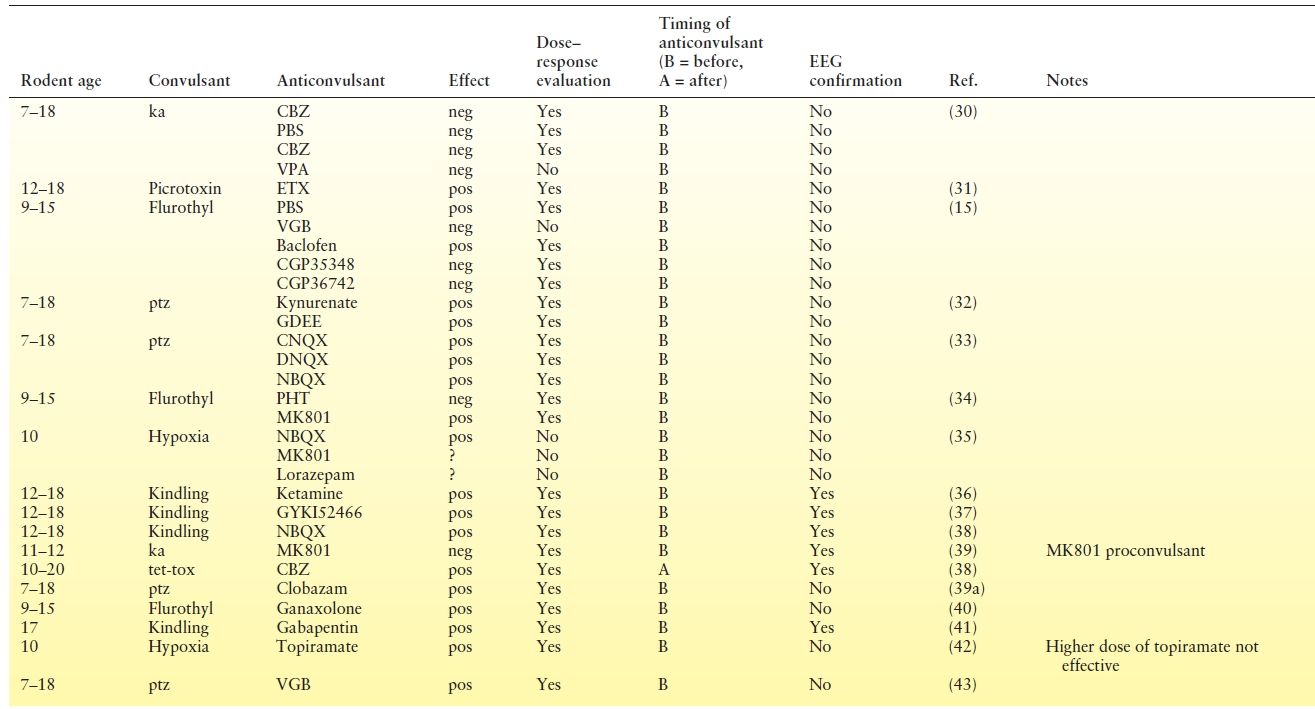
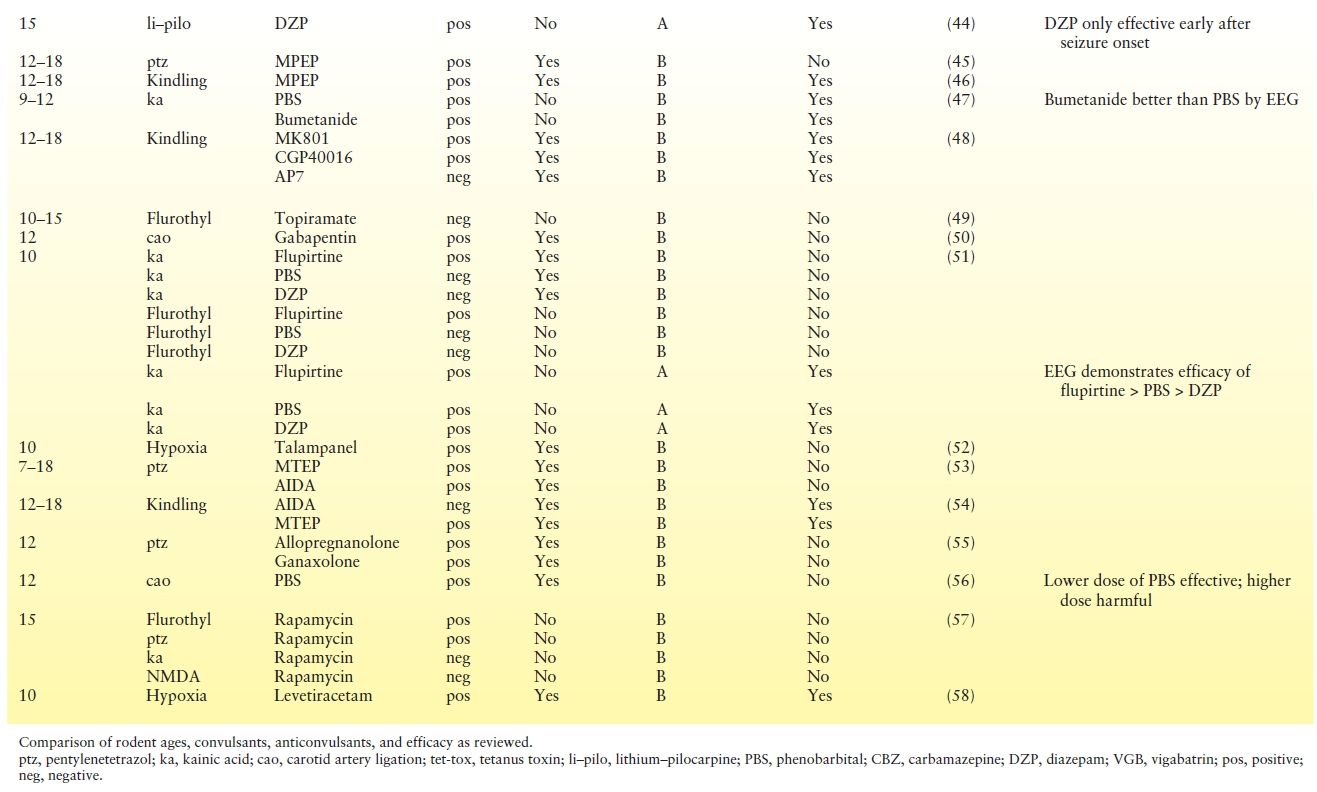
While EEG evaluation for efficacy of antiseizure medications after onset of seizures may be the gold standard, it is important to recognize the experimental issues. Continuous video-EEG monitoring for nursing pups is technically challenging. Due to the growing skull, the recording duration is limited. The numbers and locations of electrodes are limited by the size of the skull and lack of accepted standardization. Further, details of background rhythms, seizure detection, and quantification lack widely accepted quantification and standardization.
Studies in immature rodent have demonstrated that, at relevant doses, phenobarbital, phenytoin, NMDA receptor antagonists, diazepam, valproate, and vigabatrin are proapoptotic. At higher doses, lamotrigine, carbamazepine, and topiramate are proapoptotic; in contrast, levetiracetam does not demonstrate apoptosis (59). Also, phenobarbital, phenytoin, and lamotrigine have negative behavioral consequences when administered to immature rodents (60).
In conclusion, preclinical rodent studies do not offer full evidence to recommend any antiseizure medication as efficacious for neonatal seizures as observed in clinical practice. Many drugs do affect seizure threshold, as demonstrated by those studies that administered the antiseizure medication prior to the seizure. Flupirtine shows best EEG evidence; however, phenobarbital does have efficacy. Further studies are needed that do not pretreat and that use EEG, evaluate the dose response, and fully consider short- and long-term outcomes.
MECHANISMS OF SE AND EPILEPTOGENESIS
Mechanisms of SE
Here, there are two basic questions: Why did the seizure not stop by itself, and why is SE more difficult to stop with antiseizure medications than a single seizure? Was the underlying neuronal network susceptible to this happening, or did it become dynamically changed to allow its progression? Given that it has been found that the clinical situation is mimicked by the experimental in which benzodiazepines lose their potency as the seizure progresses (61), much effort has focused on the role of GABAA receptors and inhibitory synaptic transmission (62). These questions have been approached in a variety of ways, using in vitro brain slices or in vivo models employing pilocarpine, kainate, or kindling, sometimes in combination with in vitro brain slices prepared during or after the event. Recent studies suggest that during SE, GABAA receptors at inhibitory synapses onto granule cells of the dentate gyrus are removed from synaptic sites and moved to extrasynaptic sites and internal pools (63) in a subunit-specific manner (64). This likely minimizes their effectiveness in both self-termination of the seizure and the loss of effectiveness of benzodiazepines, in part mediated by loss of γ2 subunits, which modulate benzodiazepine sensitivity. These issues are complicated during development in the CA3 region of the hippocampus, where GABA-ergic synapses are depolarizing and thus contribute to the development of ictal activity (65,66); although recently the role of excitatory GABAA receptor-mediated currents has been questioned (67).
The alterations in GABAA receptors in the dentate gyrus are possibly mediated by NMDA receptor activation rather than by direct activation of GABAA receptors (63). It has been found that blocking NMDA receptors prevents the progression to drug-resistant SE (68). NMDARs then further contribute to the process as they are progressively recruited to synaptic sites as SE progresses (68). While in vitro studies suggest that NMDA receptors and AMPA receptors are involved in epileptogenesis (69–71), it is possible that their contribution to this process may be mediated by their effects on SE. Reductions in the AMPA receptor GluA2 subunit in CA1 and CA3 (72,73) 6 to 48 hours after SE, while implicated in cell death, may have also contribute to prolonging SE, perhaps through facilitated AMPA receptor function (74). Excess glutamate, which may occur with transporter dysfunction, has been shown to lead to NMDA receptor activation and seizures (75); however, this effect may be limited to developing animals in which glial regulation of extracellular glutamate by transporters is immature (76). Indeed, multiple genes, including those involved in transcription, are likely regulated following SE (77). Recent work has shown alterations in calcium-permeable AMPA receptors as a potential mechanism to prolong SE (78).
Mechanisms of Epileptogenesis
Epileptogenesis refers to the process by which a previously “normal” brain becomes capable of producing SRS. Animal models have typically employed prolonged SE to trigger this process; however, models of trauma and injections of toxins have also been used (see Review of Techniques). The nature and mechanisms of this process have been richly studied. Does this happen quickly or gradually, that is, what is the significance of the latent period between trigger and first SRS? This question is critical as it might represent a window of opportunity for intervention. What is the relationship of the sclerotic pathology, often seen in human TLE and animal models, to this process? How much of the process is due to network rewiring versus changes in neuronal and/or synaptic function? What are the signaling cascades mediating these processes and how can they be circumvented or reversed? Activation of group 1 metabotropic glutamate receptors, by themselves, has been shown to be epileptogenic (79,80), suggesting that the process might be less complicated.
The appearance of SRS has been taken to indicate the end of the latent period. Enhanced excitability has been shown to gradually develop prior to the appearance of SRS (81), suggesting the end of the latent period is not a stepwise function into SRS and epilepsy. In support of this, an intensive video-EEG monitoring study has challenged the notion of the latent period by showing that the progression into SRS and epilepsy is a sigmoid function of time (82). In other words, after the first SRS, epilepsy continues to progress. Progression clearly represents a worst-case scenario, which may not always be present (83). Additional work is needed to determine where and if there is a window for interventions to prevent this progression.
Plasticity and Trafficking of GABAA Receptors in Epileptogenesis
During the process of epileptogenesis in animal models, there are alterations in the expression and membrane localization of several GABAA receptor subunits (α1, α4, γ2, and δ) in hippocampal dentate granule neurons (84–86). These alterations, which are associated with changes in phasic and tonic GABAA receptor-mediated inhibition, and in GABAA receptor modulation by benzodiazepines, neurosteroids, and zinc, begin soon after SE and continue as animals become epileptic (84–87). Several laboratories have documented similar changes in GABAA receptor subunit composition in human TLE and in animal models of TLE (84,86,88,89). Following pilocarpine-induced SE in adult rodents, GABAA receptor α1 subunit mRNA expression decreases and α4 subunit mRNA expression increases in dentate granule cells (DGCs) of the hippocampus, and animals uniformly go on to develop the recurrent spontaneous seizures that define epilepsy (84). The change in subunit expression correlates with a decreased sensitivity to zolpidem augmentation and increased sensitivity to zinc inhibition of GABAA receptor responses (84). Similar functional and subunit expression changes have been observed in DGCs isolated from surgically resected hippocampus from patients with intractable TLE (89). The changes in GABAA receptor subunit expression and function in DGCs of adult epileptic animals precede the development of epilepsy, and immature animals exposed to prolonged induced seizures show increased GABAA receptor α1 subunit expression and do not subsequently develop epilepsy (90), suggesting that GABAA receptor changes contribute to the epileptogenic process. Viral gene transfer studies demonstrating that the expression of higher α1 subunit levels inhibits development of epilepsy after SE provide further supporting evidence (91). Changes in GABAA receptor subunit expression associated with epileptogenesis vary by region and are distinct in CA1 compared to dentate gyrus (92). In addition, changes in surface expression (92) and membrane localization (93,94) of GABAA receptors related to altered receptor composition as well as loss of receptor anchoring proteins including gephyrin and GRIP (92) may play an important role in both genetic and acquired epileptogenesis.
Network Reorganization
Network reorganization in the hippocampus has been extensively studied as one of the presumed origins of SRS and because of similar findings in human TLE. Primarily, this has focused on the output of dentate granule cell neurons and has been thoroughly reviewed (8,95,96). Excitotoxic loss of mossy cells (97) in the dentate gyrus may lead to sprouting of dentate axons, known as mossy fibers (MF). The sprouted mossy fibers make aberrant excitatory connections locally in the dentate gyrus and distantly in CA3 creating an abnormal excitatory feedback circuit (98). These abnormal connections are dysfunctional, with a higher probability of activation, a larger NMDAR component (99,100), and recruitment of kainate receptors (101). These disturbances, coupled with permanent alterations in GABAA receptors (see below), are thought to result in a circuit prone to trigger seizures in other regions, such as CA3 (95). Not without controversy, MF sprouting has not been proven to be either necessary or sufficient for development of TLE [reviewed in (7,102)]. Further aberrant circuits have also been described originating in CA3 (103) and CA1 (104–106). In trauma-induced epilepsy, aberrant connections are formed in the region of injury as well as the hippocampus [reviewed in (6,26,27)]. In the region of injury, discrete regions of apical dendrites have a selective overabundance of excitatory synaptic inputs and connectivity (107,108), which with alterations in membrane voltage-gated channel properties (109) may also contribute to the epileptic state.
Excitotoxic cell loss (which may occur following SE or other insults) throughout the hippocampus is thought to be mediated by glutamate toxicity via AMPA receptors (73,109) and NMDA receptors (110).
Secondary reactive gliosis may also contribute to synaptic dysfunction (111,112). Loss of hilar mossy cells and other neurons mediating inhibition is thought to be a critical potential contributor to the hyperexcitable steady state of the epileptic hippocampus. SE also has the paradoxical effect of inducing neurogenesis in the dentate gyrus (113). Some of the newly formed neurons may also participate in MF sprouting or other aberrant circuitry that leads to the epileptic hippocampus (114), although the exact role of newborn neurons in epileptogenesis continues to be studied.
The role of network alterations and other causative phenomena in epileptogenesis appears to be differentially regulated depending on when in development the process is initiated. Kainate-induced SE in adult animals causes, over time, SRS, CA3 cell loss, MFS into CA3 and dentate gyrus, sprouting into CA1 stratum pyramidale and stratum radiatum, and impaired learning in memory tasks (104,115,116). Similar results are found with the pilocarpine model (8,116,117). However, when animals younger than 14 days are treated with either kainate or pilocarpine, the animals do not develop spontaneous seizures (see below) (90,118,119). Single or repetitive episodes of SE in infancy caused by pilocarpine are not benign, however, and have been associated with long-term abnormalities of inhibitory neurotransmission (89). Further, single or multiple episodes of SE induced by pilocarpine at postnatal day 14 or later do result in SRS (120–122) as well as deficits in memory and learning that are inconsistently associated with cell loss and/or MFS (120,122–125). Studies in other models have not provided additional clarity regarding the association of cell loss and MF sprouting to the development of epilepsy after early-life seizures. Early-life focal administration of tetanus toxin results in a chronic epileptic state that includes memory impairment without cell loss (22) but does involve MF sprouting (126). In contrast, repetitive flurothyl seizures in early development result in MF sprouting, but they do not apparently result in SRS, only a reduced seizure threshold (127–129). Chronic perforant path kindling is associated with cell loss in the dentate gyrus (130). Similarly, in prolonged temperature-induced seizures, MF sprouting gradually develops; however, reduction in seizure thresholds is seen much earlier, and SRS have been reported only infrequently (131–133). Furthermore, MF sprouting appears in a model of early-life stress, apparently unrelated to seizures (134).
Seizure- or SE-Induced Alterations in Ion Channels
Early studies of in vitro brain slice models indicated that alterations in NMDA receptors with the successive prolongation of seizure-like discharges correlated with epileptogenesis (69–71). The mechanism of non–NMDA receptor-mediated calcium influx via calcium-permeable AMPA receptors is also thought to underlie cell death in adult models of seizures (72,135–137) and hypoxia (18). AMPA receptor GluA1 subunit up-regulation has only been found in an adult model of electroconvulsive therapy (138). As noted previously, AMPA receptor GluA2 subunit “knockdown” studies have shown that down-regulation of this receptor subunit can lead to seizures and hippocampal injury (24). Clinical evidence from pathologic studies might support up-regulation of GluA1 expression (139–141). Seizures or SE in developing animals have found either no change (109,142) or a down-regulation of GluA2 (18,143) expression with no changes in GluA1 (144). Recurrent episodes of kainate-induced SE in developing animals are associated with a decrease in kainate binding (a reflection of AMPA receptors as well as KA receptors) in CA3 but not CA1 (119). Recurrent flurothyl seizures in developing animals have shown a long-term reduction in the expression of NMDA receptor subunits and the scaffolding protein PSD-95 (145). Transient alteration in the properties of synaptically activated AMPARs consistent with calcium-permeable AMPA receptors following hypoxic seizures in developing animals has been postulated to mediate the cascade resulting in later-life alterations in this model (146). These later-life alterations following the hypoxic-seizure cascade result from activation of the mammalian target of rapamycin (mTOR) pathway (58). Seizures induced by kainate in infant rats result in altered long-term potentiation, long-term depression, kindling, and learning associated with enhanced inhibition in the dentate gyrus (147) and mechanistically linked to reduced expression of the NMDA receptor GluN2A subunit, altered trafficking of AMPA receptor GluA1 subunit, and increased expression of PSD-95 (142). In contrast to hypoxia-induced seizures, early-life kainate seizures may not necessarily acutely involve the mTOR pathway; however, long-term consequences involve alterations in fragile X mental retardation protein–mediated signaling (148).
In adult epileptic animals induced by pilocarpine SE, GABA-ergic signaling is altered by specific reduction of GABAA receptor α1 subunits and an increase in α4 subunits in the dentate gyrus, resulting in a reduction in benzodiazepine sensitivity and enhanced inhibition by zinc (84). [This contrasts markedly to the developing hippocampus where pilocarpine SE does not result in epilepsy but results in an up-regulation of α1, overall receptor numbers and enhanced benzodiazepine sensitivity (90).] Changes in GABAA receptor subunit expression associated with epileptogenesis vary by region and are distinct in CA1 compared to dentate gyrus (92). In addition, changes in surface expression (92) and membrane localization (93,94) of GABAA receptors related to altered receptor composition as well as loss of receptor anchoring proteins including gephyrin and GRIP (92) may play an important role in both genetic and acquired epileptogenesis. Altered functions of voltage-gated sodium channels (149,150), T-type calcium channels (151,152), and potassium channels (153) have been described in epileptic animals and are thought to contribute to the epileptic state. In the hyperthermia model of febrile seizures, a single prolonged seizure results in permanent susceptibility to convulsants and enhanced in vitro kindling, mechanistically linked to enhancement of the voltage-gated potassium channel HCN (132,154,155).
The signaling pathways that regulate the plasticity in ion channel expression during epileptogenesis are just beginning to be elucidated. For example, recent studies have demonstrated that the mechanisms that regulate differential expression of GABAA receptor subunits in hippocampus after SE include the CREB/ICER, JAK/STAT, BDNF, and Egr3 signaling pathways (156,157). Targeting signaling pathways that alter the expression of genes involved in epileptogenesis may provide novel therapeutic approaches for preventing or inhibiting the development of epilepsy after a precipitating insult.
Sequelae Beyond Seizures
In adult models of epileptogenesis associated with cell loss and/or MF sprouting, uniformly, there is learning and memory impairment when assessed with the Morris Water Maze (MWM), a behavioral test used to assess spatial, long-term memory formation (158). Altered emotionality is also noted with fear conditioning (159). Mechanistically, this impairment is thought to be mediated by the anatomical damage, as similar deficits are observed in hippocampal lesion studies not associated with seizures or epileptogenesis (160). Similarly, in immature animals, abnormalities in the MWM are associated with histologic changes following repetitive SE (123–125), repetitive flurothyl seizures (129,161), tetanus toxin (22), and hypoxia/ischemia- (162) and hyperthermia- (132,154,155) induced seizures. In models where immature animals develop SRS, there is altered emotionality (121). Furthermore, kainate insult in infancy and again later in adulthood results in more prominent memory impairment than a single insult at either time (163). In immature animals following a kainate-induced seizure, there have not been any detectable problems with the MWM or histologic changes (116,164), including an absence of MF sprouting; similar findings have been reported for repeated episodes of kainate-induced SE in immature animals (119). As adults, these animals have only subtle abnormalities in the MWM (165), and in more difficult mazes, these animals have abnormalities most consistent with defective working memory (142,147,165,166); emotionality may be unaffected (165) [but see (166)]. Thus, permanent impairments in learning and memory are more severe in animal models when associated with significant histologic abnormalities. However, significant impairments can also exist without histologic abnormalities, which possibly reflect pathology limited to abnormal synaptic function isolated to the hippocampus.
Genetic Susceptibility
Advances in genetics have allowed for several human epilepsy syndromes associated with single gene defects to be further characterized [reviewed in (167)]. Following determination of the analogous gene in mice, similar defects can be introduced through cloning techniques in order to better understand how epilepsy develops in these syndromes as well as determine which treatments might be more efficacious. Often, the nature of the genetic defect, whether it represents a gain or loss of function, is not clear until the altered resulting protein is expressed in an intact, cloned animal model. In the animal model of Dravet syndrome, genetic knock-in of human mutations in voltage-gated sodium channels (NaV1.1) results in a phenotype very similar to that seen in humans (168,169). Importantly, these studies have highlighted how the balance between excitation and inhibition is a critical modifier in this disorder (170). However, this mechanism has recently been challenged (171). Similarly, genetic knock-in of human mutations in KCNQ2 and KCNQ3 has many similarities to the human phenotype of benign familial neonatal convulsions (172). Enhanced function of T-type calcium channels in thalamocortical circuits has been postulated to mediate childhood absence epilepsy. While specific mutations in T-type calcium channels have not been determined in the human condition, specific genetic targeting of enhanced expression of T-type calcium channels in this circuit has been found to mimic the human condition (173). However, genetic knock-in of human mutations in GABA receptors associated with generalized epilepsy syndromes has not uniformly resulted in phenotypes similar to the human conditions (174,175). Similarly, knock-in of human mutations in nicotinic acetylcholine receptors seen in autosomal dominant nocturnal frontal lobe epilepsy also does not reproduce features similar to the human syndromes (176). These negative results suggest not only the complexities of genetic technologies but also likely reflect basic underlying differences in rodent and human physiology, especially susceptibility to seizures and epilepsy.
SUMMARY
Animal models, despite their limitations, have advanced our understanding of the mechanisms of seizures and epileptogenesis. Specifically, substantial gains have been made in understanding the ability of the hippocampus and cortex to rewire themselves following insults to result in circuits capable of spontaneous seizures. Developmental models have shown how significant physiologic and behavioral alterations can result without obvious histologic changes. Important questions remain to be answered in further understanding the signaling pathways, genetic programs, and subsequent synaptic modifications that underlie epileptogenesis as well as the behavioral consequences of seizures. These discoveries are crucial to determine safe and effective pharmacologic targets for stopping seizures and curing epilepsy and its consequences.
Suggested Readings
Mazarati A. The best model for a cat is the same cat…or is it? Epilepsy Curr. 2007;7:112–114.
Binder DK, Scharfman HE, eds. Recent Advances in Epilepsy Research, Vol. 548. New York: Kluwer; 2004.
Pitkänen A, Schwartzkoin PA, eds. Models of Seizures and Epilepsy. Amsterdam, The Netherlands: Elsevier; 2006.
Dingledine R, Borges K, Bowie D, et al. The glutamate receptor ion channel. Pharmacol Rev. 1999; 51(1);7–61.
Nadler JV. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res. 2003;28:1649–1658.
Pitkanen A, Kharatishvili I, Karhunen H, et al. Epileptogenesis in experimental models. Epilepsia. 2007;48(suppl 2):13–20.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


