Strategy
Products
Stoichiometry
Advantages
Disadvantages
Potential applications
References
Fusion protein
(Fig. 1a)
Protein
1:1 predictable
+ Intracellular co-localization of the two proteins is guaranteed
+ Very space-efficient thus good for large genes
+ Inference of gene expression can be conducted both “upstream” and “downstream”
− One protein has to be compatible with N-terminal fusion and the other to C-terminal fusion to a second protein
− Protein stability, function, and immunoreactivity may change compared to the two separate proteins
• Optogenetics/DREADD
• Multi-gene-dependent enzyme replacement
[14]
[15]
Ribosome-skipping sequences
(Fig. 1b)
Protein
1:1 total amount but unpredictable amounts of fusion protein
+2A sequences are very short
+ Inference of gene expression can be conducted “upstream”
− The biological principles behind the ribosome skipping are not entirely elucidated, making sequence design a “trial-and-error” approach
• IPS cell generation
• Correction of multi-gene deficiency
• Optogenetics/DREADD
[19]
[20]
[32]
Internal ribosome entry site
(Fig. 1c)
Protein
Cell type-dependent, normally varying between 1:0.5 and 1:0.01
+ Inference of gene expression can be conducted “upstream”
−Stoichiometric ratio is very cell type-dependent making in vitro prediction of in vivo function difficult
• Optogenetics/DREADD
• Multi-gene-dependent enzyme replacement
[7]
[33]
Bidirectional promoter s
(Fig. 1d)
Any Pol II expressed RNA (protein or miRNA product)
Promoter and UTR dependent
+ Expression strength of each RNA can be tailored slightly independently through modification of UTR sequences
− No inference of gene expression can be conducted
− Can be difficult to implement in RNA base vectors as anti-sense RNA is produced during production
• Tet/Dox-regulated vectors
• Marker gene for quantitative in vitro applications
[22]
[24]
[25]
[34]
Dual promoters
(Fig. 1e–g)
Any coding or non-coding RNA (Pol II or Pol III promoter driven)
Promoter and UTR dependent
+ Each expression cassette can be tailored individually
+ Pol II and Pol III promoters can be mixed
− Risk for promoter interference
− No inference of gene expression can be conducted
• CRISPR/Cas9 in situ genome editing
• Tet/Dox-regulated vectors
• Enzyme replacement
[8]
[23]
[25]
[26]
[27]
2 Materials
Descriptions of the components in this chapter are mainly theoretical and described as a basis for in silico cloning and vector design. For the application in a specific vector design and generation of final plasmids, we refer to other descriptions of molecular cloning techniques, e.g., Molecular Cloning : A Laboratory Manual by Green and Sambrook. However, of note is that multiple approaches described in this chapter are very sensitive to frame-shifts and thus require verbatim cloning. Therefore, it is highly recommended to base the generation of fusion protein and ribosome-skipping vectors (described below) on gene synthesis products or Gibson assembly [11] and not on “traditional” restriction enzyme-based cloning. As gene synthesis is rapidly becoming cheaper and quicker, this is also an attractive alternative for design of other approaches described here as well.
3 Methods
3.1 Polycistronic Vectors
Polycistronic vector designs are the most commonly used approaches to enable the expression of two different proteins from one viral vector genome. The advantage with these approaches is that they are relatively easy to design. However, one limitation is that they are limited to expressing sequences coding for proteins. Therefore, they will not fulfill all needs in vector design. In addition, they also have other limitations, specific to each polycistronic approach, that we will discuss further on in the chapter. The three approaches covered under this section are fusion protein approach, ribosome-skipping sequences, and internal ribosome entry site s.
3.1.1 Fusion Proteins
Fusion proteins (Fig. 1a) are not technically derived from a polycistronic operon as the transcribed RNA codes only for a single open reading frame, albeit translated into a protein with (hopefully) all the functions of the two proteins fused together. However, it is the simplest way to achieve nearly the same thing as the two polycistronic approaches described below and is therefore a natural place to start.
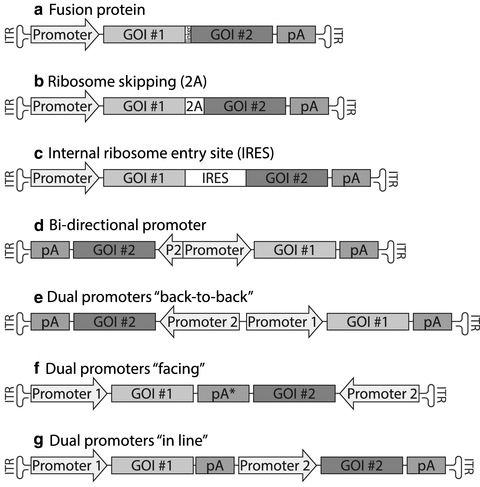
Fig. 1
Alternative designs for the expression of two proteins from one AAV vector. Schematic drawings showing the seven different design options applicable when the goal is to express two proteins (GOI #1 and #2) from the same viral vector. In this case the examples show an adeno-associated viral (AAV) vector design. The expression cassette is flanked by two inverted terminal repeats (ITR) enabling the packaging and complementary strand synthesis of the single-stranded DNA in the virus . Details on each of the seven constructs are found in the main text. Abbreviations: GOI gene of interest, pA poly-adenylation sequence, pA* a pA sequence with bidirectional activity, e.g., the full-length sv40 pA, 2A ribosome-skipping sequence, IRES internal ribosome entry site , P2 short promoter fragment, e.g., from the CMV
The design of a fusion protein requires some understanding of the proteins involved with regards to their structure, signaling/trafficking peptides, and requirements to interact with other cellular components such as cell membrane or the mitochondria. The first criterion for a successful fusion protein is that they can reside in a location in the cell compatible with the function of both proteins, i.e., a cell-surface receptor with trans-membrane domains could be compatible with a cytosolic protein if they are fused in the intracellular end of the protein while it could not be compatible with a mitochondria-associated protein.
The second step in the design of a fusion protein is to understand the optimal order between the two polypeptides. For this step there is no universal recommendation. As noted above, there may be situations where a fusion must be, say at the C-terminal of a Type I or III trans-membrane protein for the second protein to be located in the cytosol and not on the cell surface (if this is desirable). If both proteins are soluble and can reside in the same cellular compartment, e.g., two enzymes in the production cascade of a neurotransmitter, then either of the two orders may work. However, one combination may provide superior function of the complex. In some of these cases, in vitro or in vivo comparisons are required before settling on the final design.
The final component of the fusion protein is the insertion of a polypeptide linker. While a linker is not required in every design of a fusion protein, it increases the chances of success, as it is providing flexibility in the protein structure and thus will allow both proteins fused to fold in as natural way as possible.
Polypeptide linkers can be divided into three functional subgroups: flexible, ridged, and cleavable linkers. For more information see reference [12].
Applications of fusion proteins
The simplest example of a fusion protein is the addition of a short polypeptide “Tag” to either the N-terminal or the C-terminal of the expressed protein. Such tags are beneficial for a number of different functions. They are designed and evolved in close conjunction with the development of high affinity and specificity antibodies targeted toward that specific polypeptide sequence. This allows the tags to be used for recombinant protein purification or identification and visualization of the expressed protein in situ in the tissue even in cases where, either there are no available antibodies raised toward that specific protein of interest, or when you need to distinguish the ectopic expression from the endogenous, identical one.
Another interesting use of fusion proteins in viral vectors is the fusion of camelid nanobodies to RNA binding proteins or transcription factors for interaction with other proteins [13]. The development of the nanobody has risen from the unique forms of homodimeric heavy-chain antibodies (not requiring any light-chain components), to date found only in camels and related species. The variable region of these antibodies is intrinsically stable and can be truncated to form a nanobody with very high affinity to the antigen. In the paper of Ekstrand et al. 2014, they fused a nanobody (raised toward the GFP protein) to the ribosomal protein L10a and expressed it using a Cre-dependent AAV vector [14]. In vivo this was combined with a retrogradely transported canine adenovirus (CAV) to allow for a three-factor identification of dopamine cells that project to the nucleus accumbens of DAT-cre mice. Post-mortem, the nanobody-L10a fusion enabled GFP-dependent pull-down of transcribed RNA, enriched for the targeted cell population.
Fusion proteins are also commonly used in Optogenetic and DREADD studies where the channel rhodopsin or G-protein-coupled receptor is expressed using viral vectors (most commonly AAV vectors). This allows for both the identification of transduced cells in situ for live cell recording and for confirmation that the channels are correctly transported intracellularly to the site of stimulation with the optrode/ligand. The latter turned out to be a significant issue initially for the use of halorhodopsins in optogenetics [15, 16].
3.1.2 Controlling the Ribosome
In the evolutionary pressure of viral vectors to become smaller and more efficient, many viruses have developed related but slightly different approaches to achieve translation of all required virus-derived proteins from as short DNA /RNA sequence as possible. Such feats have been sometimes achieved through alternative initiation of translation of one sequence such as the VP1, 2, and 3 capsid proteins of AAV or to allow for some proteins to be expressed in trans from the same sequence that expresses a completely different protein in cis. Such approaches, while very elegant are highly sequence-dependent and are thus not universally applicable for viral vector design.
Two other approaches that viruses have developed are however much more broadly applicable, ribosome skipping and internal ribosome entry site s.
Ribosome-skipping sequences
Viruses in the Picornaviridae virus family, such as foot-and-mouth disease virus (and other related viruses) have developed a short consensus nucleotide sequence that is very unfavorable for the translation into a polypeptide sequence [17]. This sequence causes the ribosome to fail with addition of an amino acid at the C-terminal end during translation [18]. However, it does not prevent from the continued insertion of t-RNA molecule 3′ of the site. Thus, the translation continues but the new polypeptide generated is not linked to the polypeptide generated upstream of the virally derived sequence. The end result is two proteins generated from the same mRNA molecule but that are handled independently inside the cell. Such a sequence is called a ribosome-skipping sequence.
Commonly used ribosome-skipping sequences include a conserved 2A-like motif and will here collectively be referred to as just 2A sequences (Fig. 1b). The greatest advantage of 2A sequences in the application of viral vectors is that they are all very short. With a length between 21 and 30 nucleotides, they add very little to the size of the construct, something especially crucial for AAV vectors, but also when you aim to overexpress more than two proteins from the same lentiviral vector.
Secondly, the 2A sequences provide the most predictable stoichiometric ratio between the two proteins in the vector, around 1:1. However, this should not be taken as that the ribosome-skipping event is totally predictable or that all proteins generated are split into two. Neither of these is the case. The cleavage with a 2A sequence will ever be 100 % and there will always be a fraction of the protein residing as an uncleaved, fusion protein. The fraction of all protein that will be uncleaved is dependent both on the sequence the 2A is inserted into and the combination of cell type and 2A sequence utilized. Not enough is known about the 2A sequences to make this universally predictable and thus validation of each new construct is required inside the correct cell type.
1.
Applications of ribosome skipping: One of the most popular current applications of 2A sequences is the utilization in single vector-mediated reprogramming of somatic cells into IPS cells using the four Yamanaka factors, Klf4, Oct4, Sox2, and c-Myc [1]. When fused in reading frame, this construct harboring three different 2A sequences E2A, P2A, and T2A inserted into a lentiviral vector expressing all genes under a single CMV promoter, this was capable of generating IPS cells from all three germ layers [19]. Interestingly, the majority of IPS cells generated contained only a single integration, showing that an approach of mixing four separate lentiviral vectors would in such a case prove potentially very inefficient.
2.
Potential issues and recommendations: While the basic concept of the 2A sequence is straightforward, some extra attention is required in the design of the constructs. The first thing to be aware of is that the 2A sequence leaves traces in the final polypeptide. The 2A consensus sequence in itself codes for Asp-Val/Ile-Glu-X-Asn-Pro-Gly-Pro. Normally, the cleavage occurs just before the last proline (Pro) leaving only a single amino acid on the N-terminal of the downstream protein while the C-terminal of the upstream protein will have seven extra amino acids [20]. Thus, for a successful outcome, the upstream protein must handle a larger C-terminal peptide without disruption of the function. In addition, there will always be a fraction of the expressed protein that will remain as a fusion protein with an 8aa linker. Such species should preferably be functional as well, but at least not detrimental or toxic for the system studied.
A second design consideration that the 2A sequences share with the fusion proteins above is that all three sequences (first and second protein and the 2A sequence) need to be in the same reading frame. Thus, it is strongly recommended to utilize either modern cloning techniques such as Gibson assembly [11] or ordering of the sequence from gene synthesis service providers.
3.
 Lentivirus Production and Purification
Lentivirus Production and Purification
 Small-Scale Recombinant Adeno-Associated Virus Purification
Small-Scale Recombinant Adeno-Associated Virus Purification
 AAV-Mediated Gene Transfer to Dorsal Root Ganglion
AAV-Mediated Gene Transfer to Dorsal Root Ganglion
 Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
 Altering Tropism of rAAV by Directed Evolution
Altering Tropism of rAAV by Directed Evolution
 Convection Enhanced Delivery of Recombinant Adeno-associated Virus into the Mouse Brain
Convection Enhanced Delivery of Recombinant Adeno-associated Virus into the Mouse Brain




Internal ribosome entry site : The internal ribosome entry site , IRES is probably the most well-known approach for the generation of polycistronic vectors and very widely utilized (Fig. 1c). As with the 2A sequences, the IRES was first discovered in viral sequences [21]. The sequence attracts the ribosome to assemble in the middle of an mRNA molecule and initiates translation at a new start codon. It is attractive for a number of reasons. The first is that the IRES is easy to clone into a vector and to get it to function satisfactory. It does not have to be aligned to any specific reading frame. All three components (the upstream and downstream proteins and the IRES) can all be in different reading frames. The second reason is that the resulting proteins are always two separate proteins and no remnants of the IRES are found on either protein. Lastly, through the expression of the protein downstream of the IRES, the presence of the protein upstream can be reasonably well inferred. However, the opposite is not guaranteed and inferring anything about the expression level of the first protein based on the second one is not advisable. The reason for this is that the activity of the IRES is very cell type- and context-dependent. Thus the stoichiometric ratio between the two proteins expressed will vary significantly. The one thing that is certain though is that the second protein will never be more expressed than the first protein (if correct Kozak sequences are used for both reading frames).
Related posts:
Lentivirus Production and Purification
Small-Scale Recombinant Adeno-Associated Virus Purification
AAV-Mediated Gene Transfer to Dorsal Root Ganglion
Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
Altering Tropism of rAAV by Directed Evolution
Convection Enhanced Delivery of Recombinant Adeno-associated Virus into the Mouse Brain
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
