Familial Temporal Lobe Epilepsies
Fernando Cendes
Eliane Kobayashi
Iscia Lopes-Cendes
Frédérick Andermann
Eva Andermann
Introduction
A positive family history of seizures and/or epilepsy is frequently observed among patients with temporal lobe epilepsy (TLE).67 These families cannot be included in only one group, however, and detailed characterization of affected family members is crucial for defining the familial epilepsy syndrome. Patients with TLE can be identified in the context of familial mesial temporal lobe epilepsy (MTLE),9,16,19,37,43 familial TLE with auditory features,34,49,50 familial partial epilepsy with variable foci (FPEVF),56,69 and generalized epilepsy with febrile seizures plus syndrome (GEFS+),23,45,55,58,66
There are two groups of familial TLE, according to the evidence for involvement of mesial or neocortical structures. The majority of affected individuals with familial MTLE have a benign clinical course, and in some families all affected members have good seizure control, much like those originally described by Berkovic et al.9,11 However, as in patients with nonfamilial MTLE, some affected family members may have poor seizure control and require surgical treatment.19,40 Although hippocampal atrophy (HA) and other signs of hippocampal sclerosis (HS) were more frequent and more severe in those patients with refractory seizures, these changes were also observed in patients with good outcome41,43 and even in asymptomatic family members (Fig. 1).42 These are strong indicators that genetic factors play a role in the genesis of hippocampal pathology in patients with familial MTLE. The genetic background in familial MTLE, however, does not suggest a more widespread structural abnormality on magnetic resonance imaging (MRI).25
Familial TLE with auditory features, also described as autosomal-dominant partial epilepsy with auditory features (ADPEAF), and familial lateral temporal lobe epilepsy (FLTLE)38 was first reported by Ottman et al.,49 and additional families have been described by the same group and by other authors.34,48,50,52,68 Seizure semiology pointed to an extrahippocampal epileptogenic area in the temporal lobes, and, characteristically, most patients had auditory auras. Patients have good seizure control, and epileptiform discharges may be observed over posterior temporal regions. No clear signs of HA have been described in different series, but abnormalities in the neocortical aspects of the temporal lobes may be present (Figs. 2 and 3).44 Molecular studies identified linkage to chromosome 10q (ch10q),49,52,68 and mutations in the leucine-rich, glioma-inactivated 1 gene (LGI-1) have been identified.34,48,50
There is no evidence to suggest that partial epilepsy with auditory features, familial partial epilepsy with variable foci, or temporal lobe variants of benign childhood epilepsy with centrotemporal spikes ever evolve into MTLE with HS; this is further evidence for a clear distinction between familial MTLE and these other familial epilepsy syndromes.
Historical Perspective
Genetic factors in epilepsy have long been recognized. Until very recently, however, only generalized epilepsies were thought to be genetic in origin, whereas focal or partial epilepsies were largely attributed to environmental factors, such as birth injuries, infections, postnatal head trauma, and brain lesions such as tumors or vascular insults.
As in the generalized epilepsies, partial epilepsies were found to fit a model of multifactorial inheritance (now termed complex inheritance), in which there is an interaction of one or more genes and environmental factors.2,3,6
In the last decade, several autosomal-dominant forms of partial epilepsy were described (reviewed by Berkovic and Steinlein13 and more recently by Andermann et al6a). These include autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE), familial MTLE, ADPEAF, FPEVF, and autosomal-dominant rolandic epilepsy with speech dyspraxia.
The first description of familial occurrence of TLE was in 1994 by Berkovic et al.; they described it as a benign syndrome with late seizure onset without a history of prolonged febrile seizures (FS) or MRI evidence for mesial temporal sclerosis (MTS). However, in subsequent FTLE series, patients had a less benign clinical course, and a high proportion of individuals with HA were described. Some of these patients required surgical treatment for their epilepsy.19,40,43 These families showed phenotypic heterogeneity in different family members, as well as between families, with respect to a history of prolonged FS, severity of the epilepsy, and presence of HA. The original series of Berkovic et al.9,11 was population based, arising from a twin study, whereas the later series were hospital based.
Recently, FTLE was included in the new proposal for classification of epileptic syndromes by the International League Against Epilepsy (ILAE), supporting it as a well-defined syndrome.22
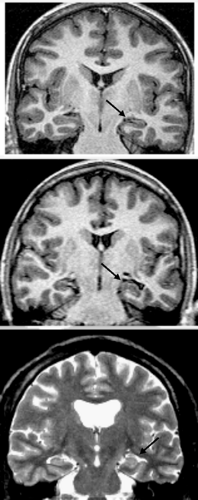 FIGURE 1. Hippocampal abnormalities in three asymptomatic individuals from families with mesial temporal lobe epilepsy. Top: Coronal T1-IR image of a 14-year-old boy, showing flattened and atrophic left hippocampus (arrow). Middle: Coronal T1-IR image from a 15-year-old boy. The left hippocampus is atrophic and bent clockwise and has an abnormal, “irregular” shape (arrow). Bottom: Coronal T2-FSE image from a 26-year-old man. The left atrophic hippocampus has an abnormal, “rounded” shape (arrow). In addition, the fusiform gyrus and collateral sulcus on the left have an abnormal shape. Note also the absence of septum pellucidum. (Reproduced, with permission, from Kobayashi E, Li LM, Lopes-Cendes I, et al. Magnetic resonance imaging evidence of hippocampal sclerosis in asymptomatic first-degree relatives of patients with familial mesial temporal lobe epilepsy. Arch Neurol. 2002;59(12):1891–1894. Copyright © 2002, American Medical Association. All rights reserved.) |
With the description of the neocortical form of familial TLE or ADPEAF49 associated with mutations in the LGI-1 gene on chromosome 10q,34,48 the distinction between mesial and lateral forms became more obvious.7,10,37,38,43,63
It is important to emphasize that it is impossible to distinguish familial from nonfamilial TLE patients based solely on the clinical presentation, in both mesial and lateral forms. Because the family history is not always accurately documented, many so-called “sporadic” or “isolated” patients may actually have a familial epilepsy syndrome.
Definitions
Familial Mesial Temporal Lobe Epilepsy
The best definition of familial MTLE is based on the familial recurrence of MTLE, according to the ILAE clinical-electroencephalogram (EEG) criteria,53 in the absence of any suggestion of other partial (including lateral TLE symptoms) or generalized epilepsy syndromes in other affected family members. Thus, the finding of at least two MTLE patients in one family is suggestive of familial MTLE. The observation of an autosomal-dominant inheritance pattern with incomplete penetrance implies the presence of asymptomatic carriers of the genetic abnormalities who can transmit the disease to their offspring. Therefore, we should consider inclusion not only of families with affected first-degree relatives, but also those with affected second- and third-degree relatives. This criterion has not been employed in some reported series, leading to exclusion of many possible familial MTLE kindreds.51
Familial Temporal Lobe Epilepsy with Auditory Features
Familial TLE with auditory features is a benign epilepsy syndrome, characterized by auditory auras (buzzing, roaring, radio- or motor-like sounds, distortions in sounds and words). Although other manifestations such as psychic, cephalic, and other sensory and motor phenomena can occur, the auditory auras are a landmark for this syndrome. Sometimes ictal aphasia and visual misperceptions can occur and, in some families, secondarily generalized tonic–clonic seizures (GTCS) are frequent.16,29,47,52,68 The pattern of inheritance observed is autosomal dominant with incomplete penetrance.
Age of onset is variable, usually in the second or third decade of life, and seizures are easily controlled by antiepileptic drugs (AEDs). EEGs may show posterior temporal epileptiform discharges but are frequently normal. No signs of HA are found on MRI studies, but a lateral temporal malformation pattern has been observed in 45% of affected individuals, including one asymptomatic carrier of the mutation.44 The left temporal lobes of these individuals seemed enlarged, and sometimes there was protrusion of the brain parenchyma laterally, with an “encephalocele-like” appearance. Anterior temporal lobe volumetry showed a significant global increase in volumes in only two individuals. The epileptogenic significance of these structural abnormalities is unknown.
Epidemiology
The prevalence and incidence of these two forms of familial TLE are unknown. However, familial MTLE is apparently more common than familial TLE with auditory features. There is no predominance in any particular ethnic group. Families with MTLE have been described in Australia, Canada, Brazil, Italy, Belgium, and France. FLTLE has been studied in the United States, Brazil, Japan, Germany, France, Italy, Spain, and Australia. This is probably an underestimate of the real prevalence of both forms of familial TLE worldwide, especially in families with predominantly good outcomes.
Ascertainment of these families requires detailed questioning of patients and family members. This has only been emphasized recently because, in the past, MTLE as well as other partial epilepsies were considered to be symptomatic and due largely to environmental factors. A preliminary hospital-based study found that familial MTLE represented 7% of all MTLE patients.43
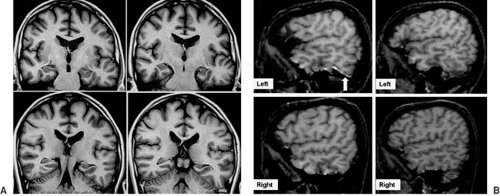 FIGURE 2. A: T1-IR coronal images from a patient with LGI-1 mutation showing left temporal lobe dysgenesis, characterized by enlargement of the lateral temporal lobe, with small gyri (although not characterizing polymicrogyria). B: T1 sagittal images from the same patient showing absence of first and second temporal sulcus on the anterior and middle portions of the left temporal lobe. The posterior basolateral aspect of the left temporal lobe is also abnormal, with a protrusion of parenchyma downward, with an “encephalocele-like” appearance (arrow). (Reproduced, with permission, from Kobayashi E, Santos NF, Torres FR, et al. Magnetic resonance imaging abnormalities in familial temporal lobe epilepsy with auditory auras. Arch Neurol. 2003;60(11):1546–1551. Copyright © 2002, American Medical Association. All rights reserved.) |
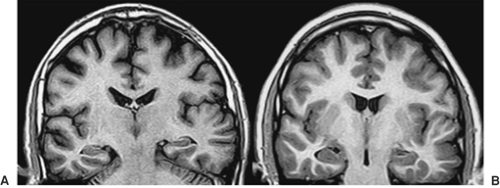 FIGURE 3. T1-IR coronal images from two patients with LGI-1 mutation, showing left temporal lobe malformation, with a dysgenetic aspect of temporal gyri and enlargement of the lateral aspect of the temporal lobe. (Reproduced with permission, from Kobayashi E, Santos NF, Torres FR, et al. Magnetic resonance imaging abnormalities in familial temporal lobe epilepsy with auditory auras. Arch Neurol. 2003;60(11):1546–1551. Copyright © 2002, American Medical Association. All rights reserved.) |
Etiology and Basic Mechanisms
The etiology of familial epilepsies is first determined by the genetic pattern that indicates an inherited disease and second
by the structural/functional abnormalities that are associated with this genetic background.
by the structural/functional abnormalities that are associated with this genetic background.
Pathogenetic mechanisms underlying familial MTLE remain unknown. Seizures and HS in familial MTLE may result from interactions between genetic and environmental factors. No locus for familial MTLE with HS has been identified.
No single-gene molecular defect has been confirmed, although several loci for familial TLE have been mapped. Digenic inheritance with loci on chromosomes 1q25–q31 and 18qter was described in a large family with febrile seizures and familial TLE without HS,8 as well as a locus on chromosome 12q22–q23.3 in another large family with autosomal-dominant familial TLE with febrile seizures without HS.21 Whether these loci represent susceptibility for febrile seizures or for TLE remains to be determined. A more recent locus was described on chromosome 4q in a four-generation kindred with familial MTLE.31 MRI data, however, did not show signs of HS in this family.31 Investigation of these loci in other families has not been reported.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


