Figure 55.1. Chemical structure of gabapentin, pregabalin, and gabapentin enacarbil.
The mechanism of action of gabapentinoids differs from that of other antiepileptic drugs (AEDs). Contrary to initial belief, they do not interact with GABA receptors. Rather, compelling evidence suggests that they bind to the α2-δ subunit of the P-, Q-, and N-type voltage-gated calcium channels (1,2). The binding affinity of pregabalin for the α2-δ subunit is six times greater than that of gabapentin (3). The functional consequences of binding to this site are yet to be fully elucidated. These may involve allosteric modulation of calcium channels (4), or inhibition of trafficking and expression of calcium channels at the presynaptic membrane (5). In any case, the ensuing effect appears to be a decrease in presynaptic calcium influx, which leads to a reduction in calcium-dependent release of neurotransmitters, such as glutamate, noradrenaline, serotonin, acetylcholine, substance P, and calcitonin gene–related peptide, from nerve terminals (6).
GABAPENTIN
Chemistry
Gabapentin is a highly water-soluble, bitter-tasting, white crystalline substance with a molecular weight of 171.34 g/mol. At 25°C, it has two pKa values at 3.68 and 10.70. At physiologic pH, it is a zwitterion—a neutral molecule with both negative and positive charges (7). In this latter form, gabapentin is recognized by the l-amino acid transport system, which mediates its transport across the gut wall, the blood–brain barrier, and cell membranes (7,8).
Pharmacokinetics
Absorption
Gabapentin is absorbed predominantly in the small intestine, where the l-amino acid transport system is concentrated (9). Absorption in the colon is poor (10).
Gabapentin exhibits dose-dependent pharmacokinetics because its oral bioavailability decreases with increasing doses (11). Oral bioavailability is approximately 60% after a 300-mg dose, but only approximately 40% after a 600-mg dose and approximately 35% at a dose of 1600 mg t.i.d. at steady state (7). Peak serum concentrations typically occur 2 to 3 hours postdose (7,9), and steady state is achieved in 1to 2 days (7). Although in phase I pharmacokinetic studies serum concentrations of gabapentin increased linearly up 1800 mg/day (12), further rises in serum concentrations were less than dose proportional at doses between 1900 and 4800 mg/day (12). A nonlinear increase in serum concentrations of gabapentin with increasing doses was also noted in clinical trials (12). Overall, these findings are consistent with the saturability of the l-amino acid transport system involved in the gastrointestinal absorption of the drug (11,13).
Absorption of gabapentin varies considerably between patients (14), resulting in substantial interindividual variations in serum concentrations (13), which may contribute to differences in dose requirements. Within subjects, however, pharmacokinetic variability appears to be relatively low (13).
No dose adjustments are required when the drug is administered with food, as this has a negligible impact on gabapentin absorption (9).
Distribution
Gabapentin is not bound to plasma proteins (15). The volume of distribution is approximately 60 L or 0.65 to 1.04 L/kg (7). Gabapentin crosses the blood–brain barrier via the l-amino acid transport system. Cerebrospinal fluid (CSF) concentrations are 5% to 35% of that of plasma (15), and after a single dose continue to rise for hours after peak serum concentrations are attained.
Elimination
Gabapentin is not metabolized in humans and is eliminated unchanged in the urine (15,16). The elimination half-life of gabapentin is 5 to 9 hours in individuals with normal renal function (17). It is unknown whether gabapentin elimination is dependent on a tubular reabsorption process. However, cimetidine (an inhibitor of renal tubular secretion) decreases gabapentin clearance by approximately 12%, suggesting that a tubular secretion process could be involved in the elimination of the drug (18).
Special Populations
Gabapentin clearance is higher in children than in adults. It is greatest in younger children, with those under 5 years of age requiring approximately 30% higher doses than do children aged 5 to 12 years (19). Gabapentin serum concentrations are higher in the elderly than in nonelderly adults receiving the same dose, a finding that is consistent with the physiologic decline in renal function (20).
Gabapentin clearance is decreased in patients with renal failure (especially in those requiring hemodialysis), potentially resulting in severe drug toxicity if appropriate dose adjustments are not made (21). Dose recommendations for different degrees of renal impairment are available in the drug information sheet (22).
New Formulations
To overcome the problems of gabapentin having a short half-life and a saturable mechanism of absorption, an extended- release formulation (Gralise) has been developed, which uses gastric-retentive technology to release gabapentin slowly over approximately 10 hours (23). This may attenuate the saturation of the l-amino acid transport system, thus enhancing and prolonging the absorption of the drug, particularly at higher doses, compared to the traditional formulation (24). Absorption of the extended-release formulation is substantially improved by food (23). Extended-release gabapentin has been found to be effective in the treatment of postherpetic neuralgia (23), but no controlled trials with this formulation have been reported as yet in patients with epilepsy.
Another agent developed to improve gabapentin absorption is gabapentin enacarbil, a prodrug of gabapentin (25). Gabapentin enacarbil is absorbed in the small and large intestine via the monocarboxylate transporter type 1 and the sodium-dependent multivitamin transporter (25). Absorption of gabapentin enacarbil is not saturable nor dose dependent, and it is improved by food. Gabapentin enacarbil is rapidly converted to gabapentin by nonspecific carboxylesterases in enterocytes and, to a lesser extent, in the liver (25). Serum concentrations of the intact prodrug are transient and represent ≤2% of those of gabapentin. The disposition of gabapentin derived from gabapentin enacarbil is similar to that of gabapentin administered as such. Gabapentin enacarbil is effective in the treatment of moderate–severe primary restless legs syndrome and postherpetic neuralgia, but no controlled studies with this agent have been reported as yet in patients with epilepsy.
Drug Interactions
Gabapentin has not been reported to cause or be a target for clinically relevant drug interactions. This is attributable to the fact that gabapentin is not bound to plasma proteins, is not metabolized, and does not induce or inhibit enzymes involved in the metabolism of other drugs (7,9).
Antacids containing aluminum or magnesium can reduce gabapentin bioavailability by approximately 20% (7), but the clinical relevance of this interaction is uncertain (7). In any case, it is generally recommended that administration of antacids and gabapentin be separated by ≥2 hours.
Efficacy
Adjunctive Therapy in Epilepsy
Adults.
Five randomized, double-blind, placebo-controlled trials demonstrated the efficacy of gabapentin as adjunctive therapy in adults with refractory partial seizures (26–30) (Table 55.1). In these studies, 600 to 1800 mg/day of gabapentin (in three divided doses) were investigated over approximately 3 months. Titration was 2 to 3 days, except for one study (26) in which the target dose was reached over a 2-week period. The proportion of patients attaining a ≥50% reduction in seizure frequency compared to baseline (responder rates) varied between 12.5% and 33% (vs. 6.7% and 16.7% with placebo), with higher doses being associated with higher responder rates. Open-label studies (31–33) and case series (34,35) have suggested additional benefit at even higher doses (up to 6400 mg/day).
Table 55.1 Randomized Placebo-Controlled Trials of Gabapentin as Adjunctive Therapy in Adults with Drug-Resistant Partial Seizures
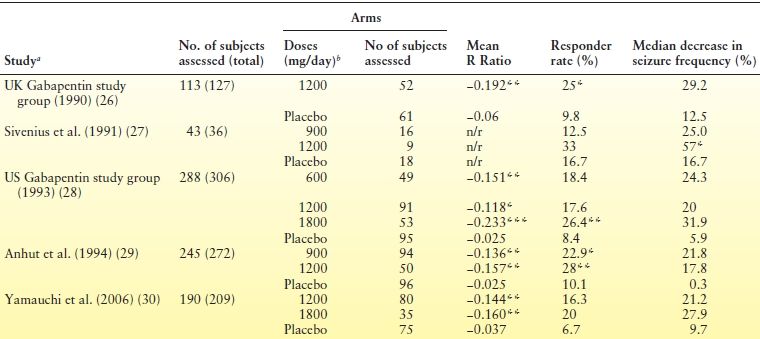
aAll studies consisted of a 12-wk baseline period, followed by a 12-wk treatment period. The only exception was the UK Gabapentin study group (26), in which the 12-week baseline was followed by a 14-week treatment period.bIn three divided doses.
*P < 0.05,**P < 0.01 and ***P < 0.001 for comparisons with placebo.
n/r, not reported; R Ratio = (T − B)/(T + B), where T is the seizure frequency during treatment and B is the seizure frequency during baseline. Therefore, negative values indicate a reduction in the number of seizures during treatment, whereas positive values indicate an increase; Responder rate, proportion of patients with ≥50% reduction in seizure frequency.
A 14-week, double-blind, placebo-controlled trial evaluated the efficacy of gabapentin as add-on therapy for refractory generalized seizures in patients with idiopathic or symptomatic generalized epilepsy (37). A total of 129 patients with a mean age of approximately 30 years (range: 13 to 62) were randomized to placebo or 1200 mg/day of gabapentin. No significant differences were found between the two groups in terms of seizure outcome. The lack of effect on seizure outcome may be a result of the relatively low dose of gabapentin used in the study.
Children.
A 12-week, randomized, double-blind, placebo- controlled trial of 247 patients provided evidence for the efficacy of gabapentin as adjunctive therapy for refractory partial seizures in children aged 3 to 12 years (38). The target dose was 25 to 35 mg/kg/day. Children receiving gabapentin had a median reduction of 35% in the frequency of complex partial seizures (vs. 12% in the placebo group) and 28% in the frequency of secondarily generalized seizures (vs. a 13% increase in the placebo group). Higher daily doses (up to 78 mg/kg/day) have been used in open-label studies, but do not appear to be associated with increased efficacy (39).
The efficacy of gabapentin (40 mg/kg/day) as add-on treatment for refractory partial seizures in very young patients has been investigated in a 3-day, randomized, double-blind, placebo-controlled trial in 76 patients aged 1 to 36 months (40). Although there was a reduction in seizure frequency in the gabapentin arm versus a worsening in patients receiving placebo, differences did not reach statistical significance.
Monotherapy in Epilepsy
Adults.
The efficacy of gabapentin as monotherapy for refractory partial seizures has been evaluated in two randomized double-blind trials (41,42). One study compared 300 and 3600 mg/day over an 8-day period in 82 hospitalized patients whose medications had been discontinued for seizure monitoring (41). Time to meet exit criteria primarily related to lack of efficacy (the primary outcome variable) was significantly longer and completion rate was significantly higher in the 3600 mg/day arm. In the second study, 275 patients were randomized to 600, 1200, or 2400 mg/day and then underwent gradual discontinuation of their concomitant AEDs (42). Duration of the double-blind treatment phase was 26 weeks (2 weeks of gabapentin add-on therapy; 8 weeks of gradual discontinuation of concomitant AEDs; and 16 weeks of gabapentin monotherapy). Outcome measures, including time to exit, completion rate, and mean time on monotherapy, did not differ across the three groups. Overall, completion rate was only 20%.
Several studies assessed the efficacy of gabapentin monotherapy in adults with newly diagnosed seizures. Chadwick et al. (43) randomized 275 patients with newly diagnosed epilepsy to one of three masked doses of gabapentin (300, 900 or 1800 mg/day) or an open-label fixed dose of immediate-release carbamazepine (600 mg/day). After titration (7 days for gabapentin, 21 days for carbamazepine), patients entered a 24-week evaluation phase. Completion rate was 37% in the carbamazepine arm and 25%, 39% and 38% in patients receiving 300, 900 and 1800 mg/day, respectively. Although the completion rate for 900 and 1800 mg/day of gabapentin was similar to that of carbamazepine, more patients in the two gabapentin groups (40% and 43%, respectively) exited the study due to seizure occurrence compared to the carbamazepine group (30%). Conversely, the withdrawal rate because of adverse events was higher with carbamazepine compared to gabapentin 900 and 1800 mg/day (24% vs. 4% and 14%, respectively). In a 30-week, randomized, double-blind study, Brodie et al. (44) compared 1200 to 3600 mg/day of gabapentin to 100 to 300 mg/day of lamotrigine in 309 patients with newly diagnosed partial or generalized epilepsy. No between-group differences were found across several outcome measures. However, since many patients had only a few seizures in the ≥12 months prior to enrollment (median seizure number at study entry: 3 for the gabapentin group and 4 for the lamotrigine group), a period >30 weeks would have been desirable to obtain a more robust assessment of comparative efficacy. In an open-label trial (45), 1721 patients for whom carbamazepine was deemed to be preferable to valproate as initial treatment were randomized to carbamazepine (100 to 2000 mg/day), gabapentin (300 to 4800 mg/day), lamotrigine (20 to 800 mg/day), topiramate (25 to 600 mg/day), and oxcarbazepine (300 to 2850 mg/day), and followed for up to 6 years. Most patients (82%) had newly diagnosed seizures, but patients who had previously received suboptimal treatment or who had relapsed after discontinuation of effective therapy were also included. Time to treatment failure, one of two primary endpoints, was significantly better with lamotrigine compared to gabapentin (hazard ratio 0.65 [95% CI 0.52–0.80]). Time to 12-month remission, the other primary endpoint, significantly favored carbamazepine over gabapentin (0.75 [0.63–0.90]).
A randomized, double-blind, parallel study compared gabapentin (target dose: 1500 mg/day), lamotrigine (150 mg/day), and immediate-release carbamazepine (600 mg/day) in 593 elderly patients (mean age 72 years) with newly diagnosed seizures (36). Patients were followed for up to 2 years. Doses could be optimized on the basis of clinical response. Although carbamazepine was the most efficacious agent, significantly more patients in the lamotrigine and gabapentin groups (55.8% and 49%, respectively) remained in the trial for 12 months, the primary outcome measure, compared to the carbamazepine group (35.5%; P < 0.0001 for lamotrigine and P = 0.008 for gabapentin vs. carbamazepine). This was largely attributable to the fact that carbamazepine was associated with higher withdrawal rates due to adverse events (31% vs. 12.6% for lamotrigine and 21.6% for gabapentin; P = 0.001).
Children.
The efficacy of gabapentin (9.7 to 19.1 mg/kg/day) in newly diagnosed childhood absence epilepsy has been investigated in two identical 2-week, randomized, double-blind, placebo-controlled trials including 33 children aged 4 to 12 years (46). Gabapentin did not significantly improve or worsen seizure frequency compared to placebo.
A double-blind study suggested that gabapentin monotherapy may be efficacious in benign childhood epilepsy with centrotemporal spikes (47). A total of 225 patients aged 4 to 13 years were randomized to gabapentin (30 mg/kg/day) or placebo for 34 weeks. In preliminary analyses, time to treatment failure was significantly longer with gabapentin compared to placebo. The estimated completion rate was 57% in the gabapentin arm and 44% in the placebo arm. These preliminary data, however, have not been followed by a final report.
Nonepilepsy Indications
There is evidence from randomized, double-blind, placebo- controlled trials that gabapentin is effective in the treatment of neuropathic pain disorders, particularly postherpetic neuralgia (48). Other conditions for which gabapentin may be useful include restless legs syndrome, anxiety disorders, and postoperative pain (49–51).
Adverse Effects
Gabapentin is generally well tolerated. In early trials, dropout rates due to adverse events were <10% in patients receiving gabapentin. Its adverse effects typically involve the central nervous system (CNS), such as drowsiness, fatigue, dizziness and ataxia (Table 55.2), tend to be mild to moderate in severity, often appear in the first few days of therapy, and may resolve within 2 to 3 weeks (13). These effects do not display a clear dose–response relationship (31,33) (see Table 55.2), with some individuals not tolerating even small doses of the drug (33).
Table 55.2 Most Common Treatment-Emergent Adverse Events in a Placebo-Controlled Trial of Gabapentin Versus a Placebo-Controlled Trial of Pregabalin
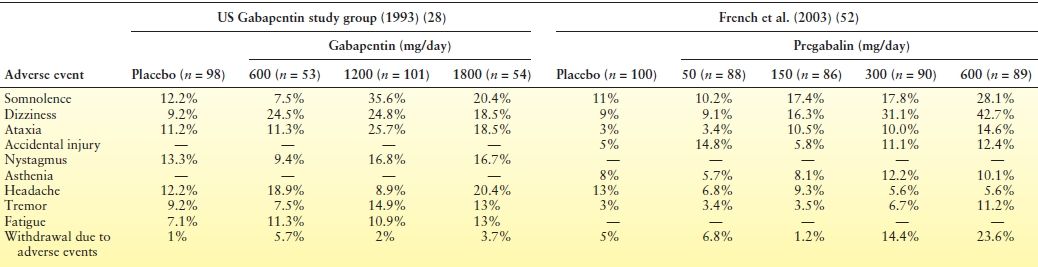
Note that, for both gabapentin and pregabalin, adverse events mainly involve the CNS. However, while gabapentin adverse events do not clearly display a dose-dependent relationship, those emerging during pregabalin therapy appear to be largely dose related.
A relatively common complication of gabapentin therapy is weight gain. DeToledo et al. (53) reviewed changes in body weight in 44 patients with refractory seizures receiving gabapentin for ≥12 months. Twenty-eight patients were taking doses of >3000 mg/day. Overall, 10 patients gained >10% of their initial weight, 15 patients gained 5% to 10%, 16 patients had no change, and 3 patients lost 5% to 10%. Gabapentin-induced weight gain appears to be dose related. In a 6-month open-label study of gabapentin add-on therapy for refractory partial seizures, weight gain was observed in 8%, 6.3%, 9.7%, and 15.2% of patients receiving 1200, 1600, 2000, and 2400 mg/day, respectively (31).
Gabapentin can cause peripheral edema, particularly at higher doses. In a pooled analysis of three randomized, double-blind, placebo-controlled studies of gabapentin in postherpetic neuralgia, the incidence of peripheral edema was 1.6%, 1.4%, and 7.5% in patients receiving placebo (n = 245), <1800 mg/day (n = 358), and ≥1800 mg/day (n = 321) of gabapentin, respectively (54). The edema resolves with discontinuation of therapy.
Movement disorders have been observed with gabapentin (55). There are several reports of gabapentin-induced myoclonus, for which individuals with preexisting myoclonus, severe chronic static encephalopathy, or renal failure appear to be at higher risk (56,57).
A few reports suggest that, in children and individuals with intellectual disabilities, gabapentin may induce behavioral problems, such as hyperactivity and aggression (51).
Serious idiosyncratic reactions are extremely uncommon during gabapentin therapy, and the potential for causing hypersensitivity reactions is much lower with gabapentin compared to some other AEDs, such as carbamazepine, phenytoin, and lamotrigine (58,59). Of note, three reports suggested that gabapentin may cause rhabdomyolysis (60–62).
There is sparse information on the fetal risks in gabapentin-exposed pregnancies (Table 55.3). As reflected by the large confidence intervals in Table 55.3, the number of exposed cases in each study is too small to allow meaningful conclusions about the teratogenic potential of gabapentin. In a recent prospective study (63), rates of major malformations in the offspring were 4.1% among 223 pregnancies exposed to gabapentin compared to 2.5% among 223 pregnancies exposed to agents considered nonteratogenic (e.g., acetaminophen or antibiotics). Gabapentin-exposed pregnancies were associated with lower rates of live births (76.2% vs. 90%, P < 0.001), and higher rates of therapeutic abortions (13% vs. 2.2%, P < 0.001), preterm births (10.5% vs. 3.9%, P = 0.019), low birth weight (8.1% vs. 4%, P = 0.033), and admissions to neonatal intensive care unit/special care nursery (38% vs. 2.9%, P < 0.001). However, because no distinction was made between monotherapy and polytherapy, and pregnancy outcomes with other AEDs for comparison are missing, these findings are difficult to interpret.
Table 55.3 Rates of Major Congenital Malformations After Exposure to Gabapentin Monotherapy in Different Prospective Studies
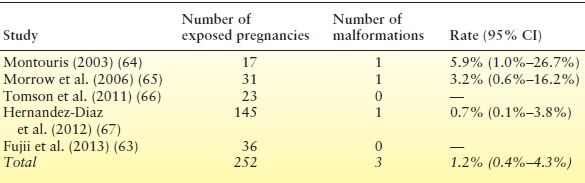
Inclusion criteria and methods of assessment (including duration of follow-up postnatally) differed across studies.
CI, confidence interval.
Place in Current Therapy
Gabapentin has been approved in several countries for adjunctive therapy and monotherapy use in partial seizures, as well as for the treatment of neuropathic pain. In the United States, it has received Food and Drug Administration (FDA) approval for adjunctive therapy in the treatment of partial seizures in patients aged ≥3 years, and for the management of postherpetic neuralgia in adults.
In the Neurontin U.S. Physician Prescribing Information (68), the recommended starting dose for patients with epilepsy aged >12 years is 300 mg t.i.d. However, if a rapid response is required, starting doses up to 3600 mg/day can be well tolerated. The effective dose range of Neurontin is given as 900 to 1800 mg/day (68). However, postmarketing experience suggests additional benefit with further dose increases, up to 3600 mg/day.
In children with epilepsy aged 3 to 12 years, the recommended starting dose is 10 to 15 mg/kg/day in three divided doses, which can be increased to the effective dose over a period of approximately 3 days (68). The effective dose of Neurontin is stated as 25 to 35 mg/kg/day in children aged 5 to 12 years, and 40 mg/kg/day in children aged 3 and 4 years (68).
Neither the extended-release formulation of gabapentin nor gabapentin enacarbil is indicated for the treatment of epilepsy. Extended-release gabapentin has received FDA approval for the treatment of postherpetic neuralgia in adults. Gabapentin enacarbil is FDA approved for the treatment of moderate–severe primary restless legs syndrome in adults, and postherpetic neuralgia in adults.
In the Gralise Prescribing Information (69), the recommended starting dose of extended-release gabapentin is 300 mg once daily, which should be increased to a 1800-mg once-daily dose over 15 days (69). Extended-release gabapentin should be taken with the evening meal. In the Horizant Prescribing Information (25), the recommended dose of gabapentin enacarbil is 600 mg once daily at 5 pm for the treatment of restless legs syndrome. For the treatment of postherpetic neuralgia, the recommended dose is 600 mg once daily for 3 days, which should be increased to 600 mg twice daily beginning on day 4 (25). When discontinuing gabapentin enacarbil, tapering is not necessary in patients taking ≤600 mg/day (25). In patients with postherpetic neuralgia taking 600 mg twice daily, the dose of gabapentin enacarbil should be reduced to 600 mg once daily for 1 week prior to discontinuation to reduce the risk of withdrawal seizure (25).
In summary, gabapentin is a safe and well-tolerated agent, which displays a wide therapeutic window and no clinically relevant drug interactions. It is also suitable for rapid titration, with little or no significant risk for serious toxicity. Although well-designed head-to-head comparative studies are lacking, gabapentin is generally considered less efficacious than other AEDs for the treatment of partial seizures, possibly due to incomplete bioavailability especially at the high doses. There is no evidence of gabapentin being efficacious in generalized epilepsies. The extended-release formulations of gabapentin and gabapentin enacarbil have yet to be investigated in patients with epilepsy.
PREGABALIN
Chemistry
Pregabalin is a white to off-white crystalline substance with a molecular weight of 159.23 g/mol. It has two pKa, 4.2 and 10.6, corresponding to the carboxylic acid and the amine groups, respectively (see Fig. 55.1). It is freely soluble in water and in basic and acidic aqueous solutions (70).
Pharmacokinetics
Absorption
Like gabapentin, pregabalin is absorbed in the small intestine by the l-amino acid transport system (9,71). Unlike gabapentin, which shows dose-dependent pharmacokinetics due to saturable absorption, pregabalin exhibits linear absorption pharmacokinetics within the therapeutic dose range (9,70). This can be explained by the fact that pregabalin is used clinically at much lower doses compared to gabapentin, and also by differences in regional distribution and sodium dependence between the pregabalin and gabapentin carriers (72). In addition, mechanisms other than the l-amino acid transport system may be involved in the absorption of pregabalin, and contribute to the >90% oral bioavailability across the effective dose range (9). Peak serum concentrations occur approximately 1 hour after oral intake, and steady state is achieved 1 to 2 days after repeated dosing. Food delays peak serum concentrations but does not affect total drug absorption (73).
Distribution
Pregabalin does not bind to plasma proteins, and it has a volume of distribution of approximately 0.5 L/kg. Pregabalin crosses the blood–brain barrier via the l-amino acid transport system. Peak CSF concentrations occur 8 hours postdose, and decrease at a slower rate compared to plasma (9).
Elimination
Pregabalin undergoes negligible metabolism (<2% of the dose) and is eliminated virtually unchanged in the urine (70,71). Its elimination half-life is 5 to 7 hours in people with normal renal function (70). Pregabalin renal clearance (67.0 to 80.9 mL/min) has been found to be lower than the glomerular filtration rate, suggesting that tubular reabsorption may be involved in the renal clearance of the drug (9).
Special Populations
Pregabalin clearance is decreased in patients with impaired renal function. This decrease is directly proportional to the reduction in creatinine clearance (CLCr) (71). A 50% reduction in pregabalin daily dose is recommended in patients with CLCr between 30 and 60 mL/min compared to those with CLCr greater than 60 mL/min. Daily doses should be further reduced by 50% for each additional 50% decrease in CLCr (74). Since pregabalin is highly cleared by hemodialysis, supplemental doses may be required for patients on chronic hemodialysis treatment after each dialysis session to maintain unaltered drug serum concentrations (74).
Preliminary observations suggest that pregabalin clearance may be also reduced in the elderly (75), a finding that can be explained by the physiologic age-related decline in renal function (76).
Drug Interactions
Since pregabalin is not bound to plasma proteins, does not influence the activity of drug metabolizing enzymes, and is not itself significantly metabolized (71), its potential for clinically relevant drug interactions is very low. In regulatory trials, pregabalin did not alter serum concentrations of concomitantly administered carbamazepine, lamotrigine, phenobarbital, phenytoin, tiagabine, topiramate, and valproic acid (77). Evidence also exists that pregabalin pharmacokinetics are not affected by several comedications, including other AEDs, oral contraceptives, oral hypoglycemics, diuretics, and insulin (78). In one study, however, comedication with enzyme-inducing AEDs was associated with moderately lower pregabalin serum concentrations compared to comedication with noninducers (75).
Pregabalin may worsen the cognitive and motor dysfunction caused by oxycodone, and potentiate the CNS effects of ethanol and lorazepam (79).
Efficacy
Adjunctive Therapy in Epilepsy
Adults.
Six randomized, double-blind, placebo-controlled trials involving 2009 patients demonstrated the efficacy of pregabalin as adjunctive therapy in adults with refractory partial seizures (52,80–84) (Table 55.4). In these studies, pregabalin doses of 50 to 600 mg/day were investigated over 12 to 17 weeks; titration was 1 to 8 days, except for one study (82) in which the target dose was reached over a 2-week period. The first three studies were designed to assess dose–response relationships by using a fixed-dose regimen (52,80,81). Doses of 150, 300, and 600 mg/day were found to be superior to placebo in reducing seizure frequency, whereas 50 mg/day was not effective (see Table 55.4). Twice-daily and three times daily dosing schedules displayed similar effectiveness. The fourth and fifth studies differed from the earlier trials in that they tested a flexible-dose regimen of 150 to 600 mg/day (82,83). In both studies, pregabalin therapy was associated with a significantly greater reduction in seizure frequency compared to placebo (see Table 55.4). The sixth study randomized 434 patients to pregabalin (300/600 mg/day), lamotrigine (300/400 mg/day), or placebo for 17 weeks of double-blind treatment divided in two phases (84). Phase I consisted of 11 weeks starting with titration (1-week for pregabalin, 5-week for lamotrigine) and concluding with doses of both AEDs fixed at 300 mg/day. Phase II was an additional 6 weeks in which patients who had seizures during phase I received a higher dose of their assigned AED (pregabalin: 600 mg/day, up-titrated over 1 week; lamotrigine: 400 mg/day, no titration). Patients who were seizure free during phase I were maintained on their assigned AED at the original dose. During phase I, there were no significant differences in efficacy measures across the three groups. Over the entire 17-week treatment period, however, pregabalin was associated with a greater reduction in seizure frequency and higher responder rates compared to placebo. Responder rates, but not seizure frequency reduction, also significantly favored pregabalin over lamotrigine (see Table 55.4).
Table 55.4 Randomized Placebo-Controlled Trials of Pregabalin as Adjunctive Therapy in Adults with Drug-Resistant Partial Seizures
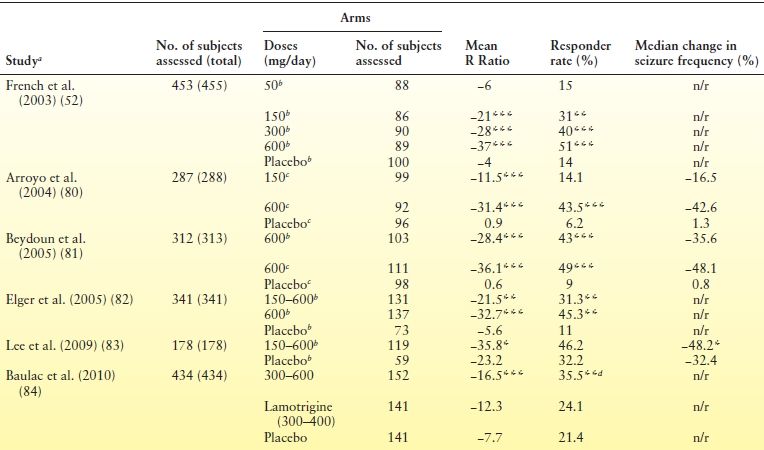
aThree studies [French et al. (52), Arroyo et al. (80), Beydoun et al. (81)] consisted of a 8-wk baseline period, followed by a 12-wk treatment period. Two studies [Elger et al. (82), Lee et al. (83)] consisted of a 6-wk baseline, followed by a 12-wk treatment period. The study by Baulac et al. (84) was characterized by a 6-wk baseline, followed by a 17-wk treatment period.bIn two divided doses.cIn three divided doses.dP < 0.05 for comparisons with lamotrigine.*P < 0.05, **P < 0.01 and ***P < 0.001 for comparisons with placebo.
n/r, not reported; R Ratio = [(T − B)/(T + B)] × 100, where T is the seizure frequency during treatment and B is the seizure frequency during baseline. Therefore, negative values indicate a reduction in the number of seizures during treatment, whereas positive values indicate an increase; Responder rate, proportion of patients with ≥50% reduction in seizure frequency.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


