Generalized (Genetic) Epilepsy with Febrile Seizures Plus
Ingrid E. Scheffer
Samuel F. Berkovic
Introduction
Generalized epilepsy with febrile seizures plus (GEFS+) is a familial epilepsy syndrome characterized by heterogeneous epilepsy subsyndromes or phenotypes within families.50 GEFS+ has been key in advancing understanding of the genetic interrelationship between epilepsy and febrile seizures. The clinical concept of GEFS+ led directly to the discovery of the role of sodium channels in epilepsy and has also highlighted the importance of γ-aminobutyric acid (GABA)A receptor subunits in causing seizure disorders.
The majority of families with GEFS+ have generalized seizure types and generalized spike-wave activity on the electroencephalogram (EEG) in addition to febrile seizures.7,9,37,45,50,54,55 Studies of families around the world have highlighted that focal seizures may also occur, leading some authors to question the nomenclature of “generalized” in generalized epilepsy with febrile seizures plus.3,7,9,30,51,58 We suggest that the nomenclature be altered to “genetic epilepsy with febrile seizures plus” to reflect this observation. It must be emphasized that generalized epilepsies are considerably more frequent in GEFS+ families.
Historical Perspectives
GEFS+ was originally described in 1997 in a large Australian family comprising many affected individuals who showed a spectrum of phenotypic severity.50 The phenotypes ranged from febrile seizures to mild generalized epilepsies to the severe end of the GEFS+ spectrum, which included myoclonic-astatic epilepsy (MAE, or Doose syndrome; see Chapter 232). This work built on the extensive clinical genetic studies of Doose, where he concluded that MAE was a polygenic disorder.17 Doose showed that family members of MAE probands most commonly had febrile and afebrile convulsions in early childhood. Evidence to support Doose’s hypothesis that MAE has a genetic basis has been gained from the conclusive findings of mutations found in some GEFS+ families, including family members with MAE.
Subsequently, the clinical spectrum of GEFS+ was further elucidated in a study of nine families, the majority of whom were identified through probands with MAE.54 The clinical genetics of GEFS+ were extrapolated further with the study of probands of families with severe myoclonic epilepsy of infancy (SMEI, or Dravet syndrome; see Chapter 230) where family members with seizure disorders had GEFS+ phenotypes.53,60
In 1998, the first gene for GEFS+ was discovered in another large Australian family. This was a mutation of the sodium channel β1 subunit gene, SCN1B,64 and was the first time that sodium channel genes were implicated in epilepsy. In 2000, Escayg et al. reported mutations in SCN1A, the gene encoding the α1 subunit of the sodium channel, in GEFS+ families,21 a finding later confirmed by other groups.3,20,30,63 The recognition that patients with SMEI, also known as Dravet syndrome, began with febrile seizures led Claes et al. to find that all seven of their patients with Dravet syndrome had SCN1A mutations.13 This work led many others to replicate the findings, highlighting the importance of SCN1A in the severe epileptic encephalopathies47 as well as in milder GEFS+ phenotypes.
The GABAA receptor γ2 subunit gene, GABRG2, has also been implicated in GEFS+ in several kindreds.7,27,62 More recently, mutations in possible “susceptibility genes” have been reported in the GABAA receptor δ subunit gene GABRD and the calcium channel subunit gene CACNA1H; these observations require confirmation.16,29
Definitions
GEFS+ is defined as a “familial epilepsy syndrome.” This syndrome is diagnosed on the basis of more than one individual within a family with a history of seizures that fits into a specific subsyndrome or phenotype of the GEFS+ spectrum (Fig. 1). It remains to be seen whether the phenotype of febrile seizures plus (see below) can be diagnosed in isolation and whether the same genes will be implicated. GEFS+ is best conceptualized as a spectrum of phenotypes seen within a family ranging in severity from mild to severe seizure disorders.
Febrile seizures (FSs) are defined as convulsive seizures with fever above 38°C that occur between 3 months and 6 years of age at their broadest limits.2
The phenotype of febrile seizures plus (FS+) refers to a number of different presentations. The most straightforward example is where febrile seizures continue past the defined upper limit of FS of 6 years. Rarely, seizures with fever may start before 3 months of age and would also be called FS+. Additionally, afebrile convulsions may occur in the setting of a child who has febrile seizures. These afebrile seizures may occur during the typical age range of FS (i.e., from 3 months to 6 years), or alternatively, they may follow on from the febrile seizures after 6 years. The afebrile convulsions may also occur after a break of several years after the last febrile seizure.
FS or FS+ may occur with afebrile generalized or partial seizures. For example, individuals may have FS/FS+ and absence, myoclonic, or atonic seizures or a constellation of generalized seizure types.50
Partial seizures emanating from the temporal or frontal lobes may also occur with FS or FS+.3,7,30,52 Although temporal lobe epilepsy (TLE) may occur in the context of hippocampal sclerosis (HS), presumably secondary to FS/FS+, TLE may also occur with normal neuroimaging and pathology and in the absence of preceding FS.51
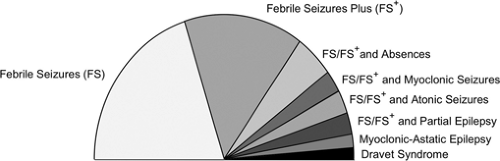 FIGURE 1. Generalized epilepsy with febrile seizures plus (GEFS+) spectrum: The spectrum illustrates the phenotypic heterogeneity seen in GEFS+ families ranging from benign phenotypes such as febrile seizures to severe epileptic encephalopathies such as Dravet syndrome. |
Epidemiology
The idiopathic generalized epilepsies (IGEs) account for about 15% to 20% of all epilepsies.28,32 The IGEs can be divided into two major subgroups: The classic IGE and GEFS+. Families in which a number of individuals have IGE typically show phenotypic heterogeneity. However, GEFS+ families show different phenotypic patterns compared with the classic IGE where childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and “generalized tonic–clonic seizures alone” segregate within families.1,42 Although overlap between the classic IGE and GEFS+ phenotypes does occur within families,41,54 this is not frequent, suggesting that these major subgroups arise due to distinct, but at times overlapping, groups of genes. No formal epidemiologic studies of GEFS+ have been performed.
Etiology and Basic Mechanisms
GEFS+ is a genetic disorder that has been recognized through study of large multiplex families where there is clinical genetic evidence of a gene of major effect. It sometimes is erroneously stated that GEFS+ is an autosomal dominant syndrome based on the initial genetic discoveries. Whilst autosomal dominant families do occur, the majority of GEFS+ families have clinical genetic evidence of complex inheritance. The rare dominant families led to the appreciation of the phenotypic variation seen in GEFS+ families and the genetic relationship between these phenotypes. However, even in these large kindreds, there is evidence for “complex monogenic” inheritance, which would explain why one family member has a benign phenotype such as FS while another has MAE or Dravet syndrome. In these latter cases, presumably other genes, with or without environmental factors, contribute to produce a severe phenotype.
Mutations of a number of ion channel genes have been identified in GEFS+ kindreds. These include sodium channel subunit and GABAA receptor subunit genes. The most frequently reported is SCN1A, which encodes the pore-forming α subunit of the sodium channel and comprises four transmembrane domains. Missense mutations spread throughout the gene have been associated with the full spectrum of GEFS+ phenotypes including MAE, Dravet syndrome, partial seizures, FS, and FS+ with other generalized seizure types.3,20,21,30,63
In contrast, SCN1A mutations usually arise de novo in Dravet syndrome and are truncation mutations in at least 50% of cases. Only about 5% of SCN1A mutations occur in patients with Dravet syndrome who have familial missense mutations where other family members have milder GEFS+ phenotypes.22,47,48 Recent reports of low levels of parental somatic and germline mosaicism explain cases of apparently de novo mutations in affected siblings; the parent may be unaffected or have a mild phenotype such as FS.15,23
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


