2
CHAPTER
![]()
Genetics
Elizabeth K. Ruzzo, Rodney A. Radtke, David B. Goldstein, and Erin L. Heinzen
Epilepsy is one of the most common neurological disorders, affecting ~3% of the human population at some period of life, with children and the elderly having the highest incidences (1). Epilepsy is a heterogeneous disorder made up of many (over 50 (2)) unique epilepsy syndromes and nonsyndromic cases. Though epilepsy has diverse etiologies, it is highly heritable and genetics play an important etiological role. A number of different lines of epidemiological evidence support this claim, including higher concordance rates in monozygotic (49%) than in dizygotic (16%) twins (3) and increased risk in first-degree relatives of probands (4–6).
Epilepsy can be broadly classified by the suspected etiology: structural–metabolic (7) (previously known as symptomatic) cases are explained by brain malformations, tumors, strokes, or other detectable phenotypes; unknown (7) (previously known as cryptogenic) cases are suspected to be explained by a clinical neurological abnormality but the cause has not been identified; and finally, genetic (7) (previously known as idiopathic) cases have no obvious cause, but are presumably genetic. Approximately 30% of all epilepsies are considered to be idiopathic and thus likely to have a genetic basis (8). Idiopathic generalized epilepsies (IGEs) have an especially strong genetic component, with ~80% concordance for monozygotic twins (9); partial epilepsies, in contrast, have a monozygotic twin concordance of 36% (9). In fact, the International League Against Epilepsy (ILAE) Commission on Classification and Terminology now refers to these cases as genetic generalized epilepsy (GGE) (7). To complicate matters further, there is evidence that some structural–metabolic cases may also have underlying genetic factors that interact with environmental factors to increase disease susceptibility (10).
Diseases explained by simple monogenic Mendelian patterns of inheritance are rare compared to complex disorders that violate Mendelian inheritance. Complex disorders are caused by interplay between genetic and environmental factors. Epilepsy is a complex disorder; however, a very small proportion of epilepsy cases, roughly 1%, show genetic transmission in a Mendelian pattern. A majority of epilepsy cases are either sporadic, with no known family history, or cluster in families that do not show a clear-cut pattern of inheritance.
OVERVIEW OF THE HUMAN GENOME
Basic Human Genetics
Deoxyribonucleic acid, or DNA, is the hereditary material in humans and almost all other living organisms. DNA is made up of four unique chemical bases, or nucleotides, including: adenine (A), cytosine (C), guanine (G), and thymine (T). This alphabet of nucleotides provides instructions for sequences of amino acids (three nucleotide combinations code for each of 20 amino acids), which the body uses to build proteins – the workhorses of the cell. A segment of DNA that codes for a protein is called a gene. The central dogma of biology states that a gene is transcribed into a messenger ribonucleic acid (mRNA) transcript, which is then translated into a protein. Before the final mRNA is translated into protein, the premRNA transcript contains both introns and exons. Introns are removed from the transcript and the exons are the portion of the gene that gets directly translated into protein.
The human genome consists of three billion base pairs of DNA and ~20,000 genes. The proportion of the human genome that is protein-coding (“the exome”) is very small, accounting for less than 2% of the genome. The remaining 98% of the genome, noncoding DNA, is not used to encode proteins. We do not fully understand the function of all noncoding DNA, however, some of it encodes RNA molecules with important biological functions (an exception to the central dogma) and other regions have important roles in regulating the expression of genes (how much gene product is available to a cell).
DNA usually exists as double-stranded DNA in which Watson–Crick base pairs (guanine-cytosine and adenine-thymine pairing via hydrogen bonds) create a regular helical structure with a sugar phosphate backbone. This double helix enables the cell to easily copy the DNA molecule during cell division, enabling precise replication of our genetic material. The DNA double helix is further organized into 23 chromosomes: 22 autosomes (1–22) and the sex chromosomes (X or Y). The human sex cells (female ova and male sperm) are haploid, meaning they contain a single copy of our genome (three billion base pairs on 23 chromosomes). Importantly, the other ~50 trillion cells in our body are diploid, meaning they have two copies of the human genome—one inherited from our mother and the other from our father. Each human diploid cell has 6 billion base pairs of DNA, and without additional modification, this DNA would be 2 meters in length. DNA is further condensed to form chromatin. DNA wraps around proteins called histones creating a series of nucleosomes (nine histone proteins + 166 base pairs of DNA) with intervening “linker DNA” of ~20 base pairs; when viewed with an electron microscope, this gives the appearance of beads on a string. Thus, nucleosomes are the structural unit of chromatin and further coiling generates tightly compacted higher-order structures. Chromosomes are most highly compacted during metaphase (a phase in mitosis of the cell cycle) and metaphase chromosomes can be viewed under a light microscope.
Finally, in addition to the chromosomal genomic DNA (gDNA), which resides in the nucleus of the cell, humans also harbor a small amount of mitochondrial DNA (mtDNA). As suggested by the name, this DNA resides in the mitochondria of the cell. Mitochondria produce energy for the cell and humans have hundreds to thousands of mitochondria per cell. Mitochondrial DNA contains 37 genes, packaged in a single circular chromosome; mtDNA is maternally inherited.
Genetic Variation in the Human Genome
When we compare the human genome to that of our closest living relatives, the chimpanzees, we see differences in only ~1% of our genomes. When we compare the genomes of human individuals, we are ~99.9% identical. So why are individuals so unique? One of the main reasons comes from genetic variation. Even that 0.1% difference means we have many different changes in our genomes. In our 3 billion base pair genome, we have ~3.5 million sites (11) (or loci) where there is a single base pair change; these sites are different from the reference genome and are known as single nucleotide variants (SNVs). Some SNVs explain differences in our physical features, others are related to disease or drug responses (see the section Pharmacogenetics), but the majority of SNVs have no known phenotypic consequences. In addition to SNVs, there are other types of genetic variation, including small insertions or deletions (indels), copy number variants (CNVs), and larger structural variants (Figure 2.1).
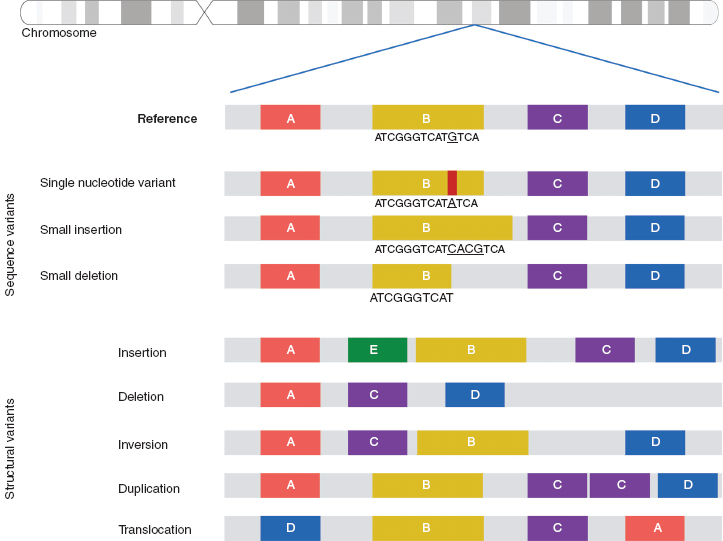
FIGURE 2.1 Types of variation in the human genome.
![]()
Each genetic variant is found at a certain frequency in the human population, with different subpopulations/ethnicities often having different frequencies for the same variant (population stratification). Typically, this frequency is described by the minor allele frequency (MAF). The MAF for a variant locus is between 0 and 50% and reflects the proportion of alleles (in the population) that are the less frequent allele (the “variant” allele). If a genetic variant is variable within or between populations, it is considered a genetic polymorphism (in contrast, genetic variants can be private). Therefore, a common single nucleotide variant is also known as a single nucleotide polymorphism (SNP). Genetic variants with different frequencies are detectable using different technologies and have unique implications for genetic analyses (Table 2.1).
OVERVIEW OF DISCOVERY GENETICS IN EPILEPSY
The initial efforts to identify genes influencing epilepsy risk came from linkage studies in rare epilepsy families with Mendelian inheritance patterns. These familial linkage studies identified over 20 “epilepsy genes” (12); however, mutations in these genes only account for an estimated 1% of epilepsy cases (see the section Early Epilepsy Genetics and Known Epilepsy Genes). This highlighted the difficulties associated with genetic discovery in a clinically and genetically heterogeneous disorder, such as epilepsy.
The next efforts came from candidate gene, and later, genome-wide association (GWA) studies. Association studies were promising because, unlike linkage analyses that rely on acquisition of multiplex families, they could be conducted in case–control populations by comparing the frequency distribution of a variant(s). The candidate gene association studies were underpowered and no convincing susceptibility genes were identified using this approach (13) (see the section Candidate Gene Association Studies in Epilepsy). Three GWA studies, completed to date, provided only modest evidence for additional loci, and replication of these signals is needed to prove their true association with epilepsy (see the section Genome-Wide Association Studies in Epilepsy).
Despite these abundant research efforts, scientists cannot explain the genetic basis of epilepsy in the vast majority of patients. The failure of association studies to discover common disease-associated variants lends credence to a rare variant-common disease model for epilepsy (14,15). Recently, the role of rare variation in human disease has become increasingly clear, especially the role of rare CNVs in neuropsychiatric disorders and epilepsy in particular (16–21) (see the section The Role of Copy Number Variants in Epilepsy). Current efforts are focused on the use of next-generation sequencing to systematically explore the role of rare variation in epilepsy (see the section Next-Generation Sequencing Studies).
Over one hundred genes have been associated with epilepsy (http://www.epigad.org; February 2014); however, the number of securely established genes is closer to 50 (Table 2.2).
Early Epilepsy Genetics and Known Epilepsy Genes
Epilepsies appearing in multiplex pedigrees with Mendelian patterns of inheritance facilitated the first genetic discoveries in epilepsy and informed our understanding of the underlying biology of seizures. In 1995, the first epilepsy-associated gene was identified in families with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (22). By studying one large ADNFLE family, a region of interest was initially identified on chromosome 20 and, subsequently, this candidate region was narrowed to a single causal missense mutation in CHRNA4, encoding the cholinergic receptor, nicotinic, alpha 4. This process is called linkage analysis, and this is a traditional technique for identifying a candidate region for a gene associated with a given disorder.
Linkage analysis makes use of crossovers, or recombination events, that occur naturally during meiosis. Linkage analysis is conducted in either a single large pedigree or across multiple smaller pedigrees with identical phenotypes. Polymorphic genetic markers (eg, SNPs or microsatellites) distributed throughout the genome are then genotyped in all individuals of the family. If the affected individuals in a pedigree nearly always inherit a genetic marker, then the disease gene and the marker are likely to be close together on the chromosome. Each marker can then be tested for co-segregation with the disease phenotype and the disease can be statistically “linked” to a specific region of the genome. In linkage analysis, a logarithm of odds (LOD) score compares the likelihood of obtaining the observed data if the tested loci are indeed linked to the likelihood of observing the same data purely by chance. If two genetic markers are on different chromosomes, then there is a 50-50 chance that they are inherited together and thus they are “unlinked.” In contrast, two genetic markers that are close together on the same chromosome have a high chance of being inherited together (“linked”), since it is less likely they will be separated by a meiotic recombination event. Thus, in linkage analysis, a “linked” marker has a high LOD score, indicating that very few meiotic recombination events have occurred between this marker and the disease gene, thus highlighting a chromosomal region of interest.
TABLE 2.1 Frequencies of Variants in the Human Population
VARIANT CATEGORY | MINOR ALLELE FREQUENCY (MAF) | IMPLICATIONS FOR GENETIC ANALYSES |
Common | 5–50% | Used in traditional GWA studies |
Less Common | 1–5% | Variants catalogued more recently and included in the newest GWA chips for association testing |
Rare | Less than 1% but polymorphic in one or more major human populations | Detectable by NGS and amenable to analysis strategies outlined in the section Next-Generation Sequencing Study Designs |
Private | Much less than 1% and found only in a single or a handful of analyzed samples and their immediate relatives | Difficult to gather statistical evidence except through co-segregation in families |
Source: Adapted from Ref. (81). Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425.
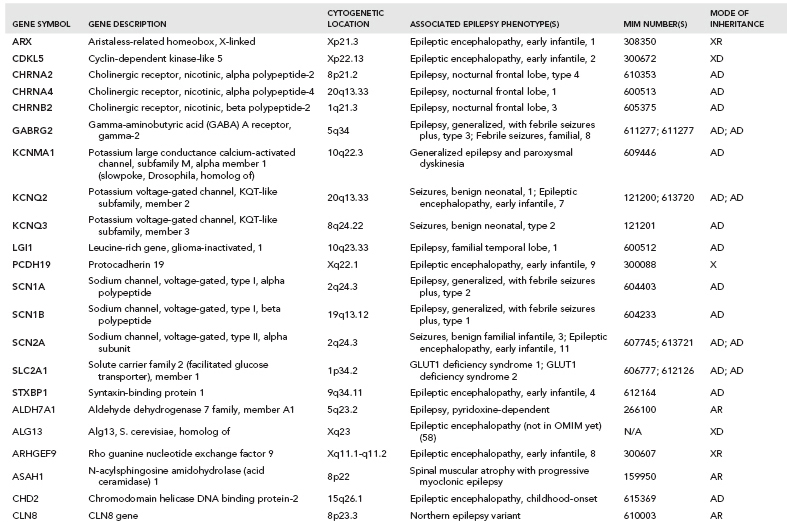
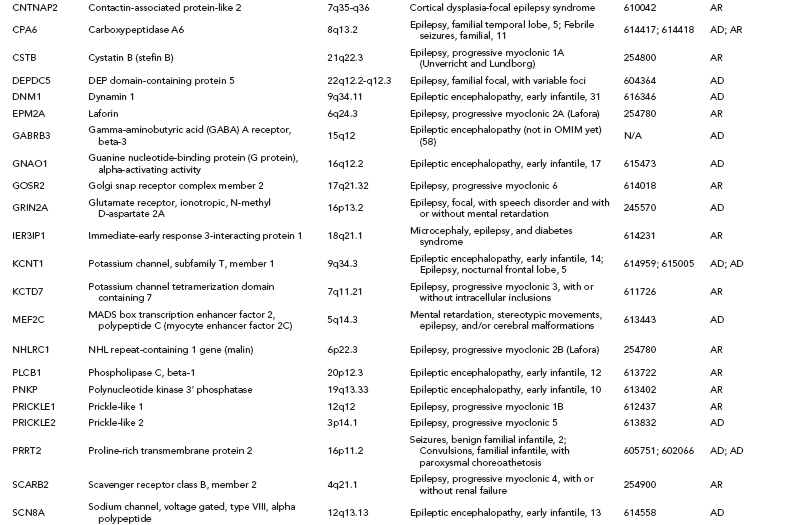
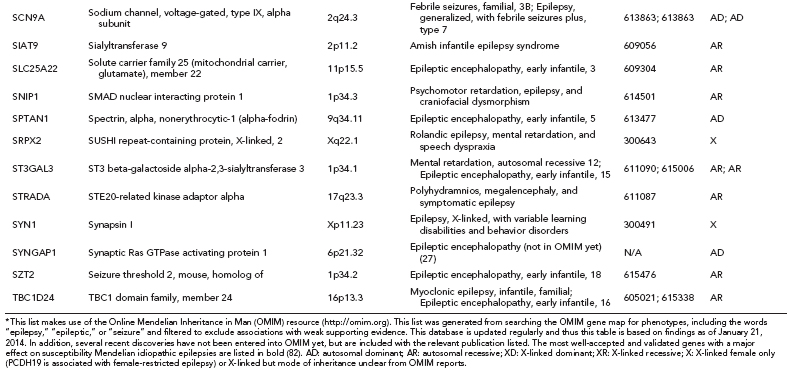
The earliest epilepsy genes fell into several main categories of voltage-gated or ligand-gated ion channels, namely subunits of acetylcholine receptors (CHRNA2, CHRNA4, and CHRNB2), subunits of sodium channels (SCN1A, SCN1B, and SCN2A), subunits of potassium channels (KCNQ2 and KCNQ3), and subunits of gamma-aminobutyric acid (GABA) receptors (GABRA1 and GABRG2). This led to the perception that epilepsy was primarily a “channelopathy,” resulting from disruption of normal electrical transmission between neurons.
While these classes of genes are still central to epileptogenesis, additional research has identified many new classes of genes (Table 2.2) highlighting the need to reconsider our narrow view of the properties of epilepsy genes. A diversity of examples exist, including: (i) leucine-rich, glioma-inactivated 1 gene (LGI1) (23), a synaptic protein that may also regulate voltage-gated potassium channels (ii) disheveled, Egl-10 and Pleckstrin (DEP) domain–containing protein 5 (DEPDC5) of unknown function (24,25) (iii) Proline-Rich Transmembrane Protein 2 (PRRT2), also of unknown function (26) and (iv) Chromodomain Helicase DNA Binding Protein 2 (CHD2) (27), a chromatin remodeling protein.
In total, linkage studies in these Mendelian epilepsy families identified over 20 “epilepsy genes” (12) (Table 2.2). Collectively, these genes explain only an estimated 1% of epilepsy cases. A majority of unsolved epilepsy cases are considered complex epilepsies, which will be explained by some combination of gene–gene or gene–environment interactions. It is also likely that the extent of genetic heterogeneity in the complex epilepsies will be greater than that of the Mendelian epilepsies, which would mean that many different genes would each explain only a very small proportion of epilepsy cases, making them evasive to current genetic approaches.
Locus Heterogeneity and Variable Expressivity
Epilepsy shows extreme genetic heterogeneity. Locus heterogeneity is evident from the relatively large number of already established epilepsy genes. A single epilepsy syndrome may be caused by mutations in one gene in family A and caused by mutations in a different gene in family B. For example, Genetic Epilepsy with Febrile Seizures Plus (GEFS+) can be caused by mutations in SCN1A, SCN2A, SCN1B, or GABRG2. Variable expressivity is also observed in epilepsy; this is when mutations in a single gene can produce different epilepsy phenotypes in different individuals. For example, mutations in SCN1A can cause GEFS+ or Dravet syndrome. Until we have fully characterized all genotype–phenotype relationships, it will be difficult to understand the shared and distinct genetic influences on different epilepsy syndromes.
Syndromes With Epilepsy as a Feature
There are two broad categories of genes associated with epilepsy: those discovered in primary epilepsy syndromes and those discovered in syndromes with epilepsy as a feature (eg, brain development disorders). Genes in the latter category have also primarily been identified by linkage analyses. Both categories still inform the biological mechanisms of epileptogenesis and provide possible therapeutic targets.
Clinically, the presentation of these syndromic and nonsyndromic phenotypes often confounds obtaining a clear diagnosis. In addition, determining the genetic basis of a patient’s epilepsy may help guide treatment and inform counseling of recurrence risk. To aid in genetic diagnoses for epilepsy, a panel of 265 genes that are “most relevant” to epilepsy have been recommended for genetic testing (28). This includes 18 phenotypic groupings or subpanels (Figure 2.2). Sequencing these 265 genes in 33 epileptic patients resulted in the identification of a presumably causal variant in 48% of patients (n = 16) (28). In addition to acting as a diagnostic tool, use of this targeted sequencing panel will also uncover the phenotypic heterogeneity associated with the less frequently mutated genes.
GENETIC ASSOCIATION STUDIES
Association studies seek to determine if two things occur together more often than expected by chance. In a classical genetic association study, these “things” are (a) a single-locus allele or genotype and (b) a phenotype (disease cases vs. healthy controls). In other words, genetic association studies can identify genetic variants that are associated with a disease or trait. If a genetic association increases susceptibility to a given disease, then the associated genetic variant will be seen more often than expected by chance in diseased individuals. While this sounds simple enough, there are many critical methodology issues to consider when conducting association studies; and true genetic associations are the result of carefully conducted studies involving large cohorts, replication cohorts, and statistically robust methods (13,29).
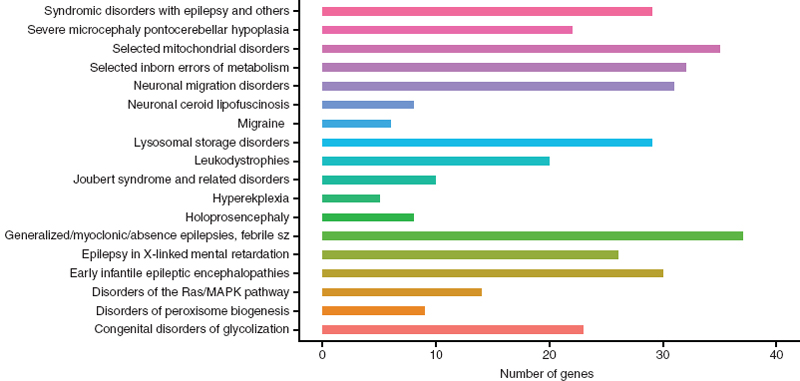
FIGURE 2.2 A bar graph of the recommended genes for NGS diagnosis in epilepsy patients categorized by phenotypic grouping/subpanel. This includes 324 unique genes; including 280 genes in one phenotypic group, 38 genes in two phenotypic groups, and 6 genes in three phenotypic groups.
Source: From Ref. (28). Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53(8):1387–1398.
![]()
Candidate Gene Association Studies in Epilepsy
Before genome-wide approaches to association studies were readily accessible, many scientists conducted association studies based on a candidate gene approach. A candidate gene may have been selected for any number of reasons including biological plausibility for the phenotype of interest. In the late 1990s and early 2000s, many genetic association studies were conducted for both focal and generalized epilepsies; in fact, over 50 studies were conducted involving hundreds of genes (13,29). The general approach of these studies was to use common SNPs as genetic markers within a given candidate gene and compare the frequency of alleles in affected cases to those in unaffected controls. These studies resulted in multiple conflicting and nonreplicable results, and ultimately failed to identify any definitive common genetic risk factors for epilepsy (13,30). One of the reasons for this inconsistency of results is the inability to accurately account for population stratification in these analyses.
Genome-Wide Association Studies in Epilepsy
Candidate gene studies came up short not only in epilepsy but also in many other disorders, and scientists recognized that a genome-wide approach was needed to obtain unbiased assessment of markers throughout the genome. Two aspects of human population genetics indicated that an “indirect approach” of assaying a set of genetic markers – even if the markers themselves had unknown functional effects – would still capture most of the common patterns of variation in the human genome and thus detect regions of the genome associated with a phenotype (31). The first relevant human population genetics observation was that approximately 90% of the genetic variants among individuals are common variants (MAF>5%) (32). The second was that the majority of common variants arose from a single mutation event that occurred on an ancestral chromosome and thus these SNPs frequently occur in combination with nearby variants.
Around the same time, two critical large-scale efforts toward understanding variation in the human genome made this vision realistic: the human genome project (draft announced in 2000) and the International HapMap project (first data release in 2003). In addition, microarray technology enabled high-throughput genotyping of hundreds of thousands of SNPs. This led to the advent of the GWA studies.
GWA studies use dense arrays of genetic markers, typically SNPs, to survey a large proportion of common variants in the human genome. SNPs are either genotyped directly or indirectly through linkage disequilibrium. Linkage disequilibrium (LD) is the nonrandom association between alleles at different loci; these loci are often in close physical proximity since the likelihood of recombination between two sites increases with the distance between them. GWA studies attempt to identify associations between genotype frequency and trait status (affected patient vs. healthy individual). The effect size describes the increased population risk for a given trait that is conferred by a given genetic variant. GWA studies have had some success in common diseases; however, the associated SNPs typically have modest effect sizes and even when all associated variants are considered collectively, they still explain only a small fraction of known heritability and thus have limited translational potential in the clinic. A current catalog of published GWA studies can be found at the National Human Genome Research Institute’s (NHGRI) website (https://www.genome.gov/26525384).
To date, three GWA studies have been conducted in epilepsy. The first study examined focal epilepsy patients of European ancestry (33). This was a phenotypically heterogeneous cohort, in that all focal epilepsies, regardless of syndrome or possible structural–metabolic causes, were included. This resulted in a cohort of nearly 3,500 cases and about 7,000 controls with no history of seizures. It is now widely accepted that the threshold for genome-wide significance in association studies is 5 × 10–8 (29); when correcting for the 528,745 SNPs genotyped in this study, the threshold required to achieve significance was 9.46 × 10–8. However, the top SNP in this study had a p-value of 3.34 × 10–7 and, thus, no SNPs were found to be significantly associated with the focal epilepsies (33). A second GWA study was also conducted in focal epilepsy patients; these patients were of Chinese ancestry (34). This GWA study was divided into two stages: a discovery stage (~500 cases (structural–metabolic focal) vs. ~3000 controls) and a replication stage (~600 cases (structural–metabolic or unknown focal) and ~500 controls). The initial discovery stage did not detect any variants of genome-wide significance. Next, they followed up a subset of SNPs with the lowest p-values (selected based on significance and regional LD structure) in the discovery stage by analyzing only these 80 SNPs in the replication stage and found one SNP that surpassed the threshold for genome-wide significance. This SNP resides on 1q32.1 in the CAMSAP1L1 gene; this gene encodes a cytoskeletal protein whose biological connection to epilepsy is unclear. It is unclear if this finding is only relevant in this ethnic population, and external replication is needed to prove the association with epilepsy. Finally, a third study was conducted in genetic generalized epilepsy (GGE) patients of European ancestry (35). Again, this GWA study was conducted in two stages: a discovery stage (~1500 GGE cases vs. ~2400 controls) and a replication stage with two independent cohorts (~600 parent–offspring trios, in which the unaffected parents were treated as controls, and an additional ~900 GGE cases and ~900 controls). No SNPs reached genome-wide significance in the discovery stage. For the replication stage, they selected a subset of SNPs with the lowest p-values in the discovery stage (selected based on significance and regional LD structure), resulting in the analysis of ~20 SNPs in the replication stage. Again, no SNPs reached genome-wide significance. Finally, they performed a combined analysis with stage 1 and stage 2 samples, and despite finding no associations of genome-wide significance, they highlight an SNP in the 5′ untranslated region (5′-UTR) of SCN1A. SCN1A has the largest number of known epilepsy-associated mutations (36) and, thus, this low signal (rs11890028, Pmeta = 4.0 × 10-6) is likely due to this SNP being in LD with rare causal mutations in SCN1A. After separating the samples into two syndromic subgroups, genetic absence epilepsies (GAEs) and juvenile myoclonic epilepsy (JME), to look for syndrome-related variants, they also found weak non-genome–wide significant signals but larger samples sizes and replication will also be needed to prove these associations.
Taken all together, these three GWA studies provided only modest evidence for additional loci and replication of these signals is needed to prove their true association with epilepsy. This is likely due to a number of different factors. The first is simply that epilepsy is a highly heterogeneous disorder and thus obtaining large cohorts for well-powered association studies requires well-phenotyped and phenotypically homogeneous samples in very large numbers. Second, it is possible that the true causal variants exist at lower frequencies in the population and thus multiple rare causal variants of large effect may be driving the diluted signals observed when assaying common variants, and thus direct identification of the causal variants may provide a better approach (37).
COPY NUMBER VARIATIONS
Deletions, insertions, duplications, and complex rearrangements of large segments of genomic DNA are all forms of structural variation (Figure 2.1). CNVs are submicroscopic structural variants. They are similar to SNPs, in that they occur throughout the human genome and confer inter-individual genetic variation. If a CNV is present in about 1% of the human population, it is called a copy number polymorphism (CNP). An SNP alters a single nucleotide pair, whereas a CNV alters anywhere from one thousand base pairs (one kilobase, Kb) to several million base pairs (megabases, Mb) of DNA. Recurrent CNVs are CNVs where the end points (beginning and end of a duplication or deletion) are limited to a narrow genomic region with extensive homology. The homology of these regions makes it more likely for these CNV events to occur and, thus, these CNVs are found in multiple individuals. In contrast, nonrecurrent CNVs have very limited homology at their end points, and thus the same end points are rarely observed in the human population.
While there are many more SNPs than CNVs in the human genome, CNVs impact a larger proportion of the genome, with at least 10% of the genome being subject to CNV (38). CNVs are found in both gene-rich and gene-poor regions. CNVs are a major genetic component of phenotypic diversity; they can be nonpathogenic and are observed in “healthy” individuals with no clinical diagnosis.
The Role of Copy Number Variants in Epilepsy
The failure of association studies to discover common variants with a clear effect in epilepsy suggested that rare variants (found in <1% of the population) might be underlying the etiology of epilepsy. Further support of this hypothesis came from a number of studies in other neurological disorders, where strong evidence emerged for rare CNVs conferring increased risk for intellectual disability (39) and schizophrenia (20). In addition, the study of rare CNVs revealed that a single CNV might be associated with a wide range of clinical phenotypes. For example, in 2008, a recurrent microdeletion at 15q13.3 was separately associated with schizophrenia (20,40), autism, and other neuropsychiatric features (41), as well as epilepsy and mental retardation (19). In 2009, this recurrent 15q13.3 microdeletion was tested in a cohort of common epilepsy patients, and found to confer increased risk for GGEs (17). This “critical region,” or minimum deleted region across all observed patients is 1.5 Mb. This 1.5 Mb region contains seven genes, including a plausible epilepsy candidate gene – CHRNA7 that encodes a subunit of the nicotinergic acetylcholine receptor. Two additional recurrent CNVs at 15q11.2 (42) and 16p13.11 (16,42) have also been associated with the common epilepsies. The microdeletion at 16p13.11 is found in GGE and partial epilepsy patients (16).
In summary, there are three recurrent CNVs that increase epilepsy risk: 15q11.2, 15q13.3, and 16p13.11. These microdeletions are especially important for the GGEs and they are also shared risk factors for other neuropsychiatric disorders, such as, schizophrenia, autism, and intellectual disability. Collectively, these three CNVs account for an estimated 2.9% of patients with genetic (a.k.a. idiopathic) epilepsies (2).
Nonrecurrent CNVs are also potential risk factors for all types of epilepsy. No single nonrecurrent CNV will account for a large proportion of epilepsy patients; however, these CNVs may include known epilepsy genes or may highlight novel candidate genes (CNVs with different end points may have a “critical region” that impacts the same novel gene). For example, in a study of ~500 epilepsy patients, two patients harbored microdeletions involving AUTS2 (previously associated with autism) and one harbored a microdeletion involving CNTNAP2 (previously associated with autism, Cortical dysplasia-focal epilepsy syndrome, and Pitt-Hopkins like syndrome 1). These are relevant to epilepsy, given the association of these genes with other neurodevelopmental and neuropsychiatric disorders (2). Investigations of CNVs in epileptic encephalopathy found that nearly 4% of patients harbor rare and clearly pathogenic CNVs (43). More generally, large heterozygous deletions (>1 Mb) are significantly enriched in epilepsy patients, and completely absent from controls when larger than 2 Mb in size (16). Proving the causality of these rare or singleton CNVs will be difficult and will likely require very large sample sizes and/or independent evidence for the candidate gene (eg, association of non-CNV mutation).
Currently, the most well-established epilepsy associated CNVs are all deletions; however, this does not to exclude the possibility that pathogenic duplications also exist.
The Mechanism of Copy Number Variant Pathogenicity
It is clear that phenotypic heterogeneity is associated with risk CNVs and it is also clear that some CNVs are not completely penetrant. The mechanism of pathogenicity for CNVs is not clear and may vary depending on the locus itself (location in the genome) or the individual genome of the patient. In many cases, a phenotype may be attributable to a single gene within the CNV; for example, a microdeletion of SCN1A in a Dravet syndrome patient. For other microdeletions, the phenotype may be simply due to haploinsufficency (only one copy of the gene does not result in enough of the gene product) of all the genes within the CNV. Alternatively, there may be a deleterious variant present in a gene on the nondeleted homologous chromosome leaving no wild type copies of the gene. Regardless of the mechanism, rare copy number variants play an important role in epilepsy susceptibility.
LARGE STRUCTURAL VARIANTS
Large structural variants (>3 Mb) can be detected by cytogenetics, which is microscopic analysis of chromosomes in individual cells. There are three main cytogenetic analyses performed in the clinical laboratory: G-banding karyotypes, fluorescence in situ hybridization (FISH), and chromosomal microarrays. Chromosomal microarrays provide the highest resolution of the three techniques and can detect submicroscopic abnormalities that are too small to be detected by conventional karyotyping (ie, CNVs).
These cytogenetically detectable variants are less frequent in the human genome than CNVs and are often pathogenic. Cytogenetic analysis is particularly helpful for epilepsies occurring with mental retardation or dysmorphic features. Eight syndromes involving seizures are caused by a recurrent chromosomal abnormality; these are referred to as the “chromosomal epilepsies” and include Down syndrome, Angelman syndrome, Miller–Dieker syndrome, Wolf–Hirschhorn syndrome (4p deletion), Chromosome 1p36 deletion syndrome, 15q inversion-duplication, Ring chromosome 14, and Ring chromosome 20 (44). In addition, Fragile X patients also frequently experience seizures. This disorder is often discussed among the chromosomal epilepsies because early research revealed that these patients frequently had a detectable fragile site on the X chromosome, at which the chromosome was prone to breakage. We now know that this disorder is caused by mutations in the FMR1 gene, most commonly an expanded CGG triplet repeat mutation. Thus, despite a commonly detectable abnormality in the X chromosome, fragile X is actually classified as a trinucleotide repeat disorder.
NEXT-GENERATION SEQUENCING STUDIES
The Human Genome Project sought to sequence the entire human genome, in other words, to determine the exact order of the base pairs in the human genome. This project used DNA from multiple anonymous volunteers and took over ten years to complete. Next-generation sequencing (NGS), also known as massively parallel sequencing (MPS), has revolutionized the cost and speed with which a human genome can be sequenced – with genomes now being sequenced within a week. While the technical details vary by the sequencing platform used, these high-throughput sequencing approaches generate millions of short sequence reads in parallel. These short sequence reads can then be aligned to the human reference genome (generated by the Human Genome Project). A computer algorithm is then used to perform “variant calling,” which results in the identification of all alleles in the newly sequenced genome that differs from the reference genome, including SNVs and indels. Additional algorithms have also been developed to identify structural variants from whole genome and exome sequence data (45–47). In exome sequencing, an additional step is included prior to sequencing, which targets and captures only the exonic and flanking intronic base pairs. Exome sequencing is popular for two main reasons: (a) a majority of known disease-causing mutations are in protein coding regions of the genome and (b) exome sequencing targets about 2% of the human genome and thus is cheaper and faster than whole genome sequencing. In contrast, whole-genome sequence data can be used to identify noncoding variants whose function we will likely understand more fully in the coming years with the advent of projects like the ENCyclopedia Of DNA Elements (ENCODE) (48).
Next-Generation Sequencing Study Designs
NGS facilitates a thorough analysis of nearly all genetic variants in the genome, including very rare variants not directly analyzed using GWA methods. These genetic variants must be prioritized differently for different diseases and study designs. At the broadest level, a number of factors should be considered, including the mode of inheritance (eg, if recessive, then focus on homozygous variants), the frequency of the disorder (eg, if the disorder is rare, the causal variant(s) will be very rare or absent in controls), and predicted deleterious nature of the variant itself. Finally, a variant-based approach could be used if the hypothesis is that causal variants will have a relative large effect and will be present in multiple cases. In contrast, if the causal variants are individually very rare in the case population but are hypothesized to lie within the same gene(s), then gene-based approaches should be used.
Research has already established that in Mendelian disease, whole genome or exome sequencing of even just a small number of cases can readily identify the causal variants as those that are shared among a small number of unrelated affected individuals and rare in the general population (49–51). In contrast, a number of different NGS study designs can be considered for optimizing discovery of disease-associated variants for complex diseases. First, a classical case–control study design can be used in which a large number of case samples and ethnically matched controls are sequenced to detect variants (or genes with qualifying variants) that are enriched in the case population. The main disadvantage to this approach is that very large sample sizes are needed, which is still cost prohibitive, to perform sufficiently powered whole-genome association studies. However, power can be increased by either restricting the tested variants based on a priori predictions of the functional impact of the variants or by sequencing individuals on one or both extreme ends of a phenotypic distribution (52).
Second, a trio-based design can be used in which the healthy biological parents and affected children are sequenced and newly formed genotypes (eg, de novo, newly homozygous) are identified in the child. The human mutation rate is between 1 × 10–8 and 2 × 10–8 per base pair per generation, which equates to roughly 40 de novo mutations per generation. Therefore, each individual is expected to have only about one exonic de novo mutation. De novo mutations, particularly those predicted to damage an encoded protein, can be disease causing and are increasingly surveyable with NGS trio studies.
Third, if families with multiple relatives affected with a complex disease exist, then family-based studies can be utilized. While any number of relatives could be selected for sequencing, one cost-effective strategy would be to sequence distantly related diseased individuals to minimize the number of variants shared by chance while still enriching for any shared risk variant(s). These shared variants can then be tested for cosegregation of the variant with affectation status in the entire family pedigree.
To date, a number of different NGS studies have been conducted with the goal of uncovering new risk factors for epilepsy and epilepsy-related syndromes. These are described further in the following paragraphs.
Case–Control Study in Genetic Generalized Epilepsy
A case–control study of 118 GGE (previously known as idiopathic generalized epilepsy) cases and 242 controls of European ancestry were exome-sequenced to evaluate the role of rare variants of relatively large effect that are frequent enough to be present in multiple cases (53). Specifically, this cohort of 118 GGE cases included 93 juvenile myoclonic epilepsy patients and 25 absence epilepsy patients. Despite restricting the tested variants to SNVs with an MAF<5% that were predicted to disrupt the protein-coding sequence, this exome-sequencing–based association testing failed to identify any variants that were significantly associated with GGE. In addition, a second stage of this study went on to genotype a subset of the SNVs identified by exome-sequencing (n = 3,897) in a larger case–control cohort of 878 GGE cases and 1,830 controls also failed to identify variants significantly associated with GGE. This work highlights the extreme genetic heterogeneity of epilepsy disorders and also suggested that gene-based analyses (as opposed to variant-based) and/or more homogeneous phenotypic cohorts are needed to reveal true risk factors for the GGEs.
Other NGS Studies in Epilepsy
There have been a handful of other NGS studies in epilepsy; primarily these have investigated specific epilepsy syndromes with familial inheritance patterns.
For example, multiple families with autosomal dominant familial focal epilepsy with variable foci (FFEVF) had previously shown linkage to 22q12, but the casual gene had yet to be identified. Therefore, two different research laboratories took the same approach; they (a) selected families with linkage to this region, (b) performed exome-sequencing on one or more family members, (c) identified rare protein-coding variants in this linkage region, and (d) tested these variants for cosegregation in the whole family. This resulted in the identification of causal mutations in DEPC5 (24,25).
Another example is benign familial infantile epilepsy (BFIE) is an autosomal dominant seizure disorder where many families showed linkage to 16p11.2-16q12.1, but the causal genetic mutations evaded discovery for many years. Therefore, one research group designed a targeted capture for genes in this linkage peak, ultimately resulting in the identification of PRRT2 as the causal gene (26). However, this discovery was in fact only due to Sanger sequencing of the PRRT2 gene because the coverage (number of short sequencing reads at a given site) was too low to accurately call variants in this gene (26). This highlights one of the technical pitfalls of NGS: coverage must be relatively high (typically >30× on average across the genome) to accurately detect variants, and this can be difficult in certain regions of the genome (eg, GC-rich).
De Novo Mutations in Epileptic Encephalopathies
The critical contribution of de novo mutations to neuro-developmental disease risk has recently been elucidated (54–56). To investigate the role of de novo mutations in epileptic encephalopathies, a large collaboration was established between the Epilepsy Phenome/Genome Project (EPGP) and the Epi4K Consortium (57). This research focused on two main types of epileptic encephalopathies, infantile spasms (IS) and Lennox–Gastaut syndrome (LGS). Exome sequencing was performed on a total of 264 probands and their unaffected biological parents (58). All putative de novo mutations were identified and subsequently validated by Sanger sequencing, resulting in an average of 1.25 de novo mutations per trio. This work identified two new epileptic encephalopathy genes and provided suggestive evidence for the role of several other genes. The two new genes both have clear statistical evidence of association with epileptic encephalopathy with four patients harboring different GABRB3 de novo mutations and two patients with the same de novo mutation in ALG13.
A number of studies have also used targeted capture to sequence only a subset of relevant genes. One such study sequenced nine known and 46 candidate genes for epileptic encephalopathy in 500 cases (27), resulting in the association of epileptic encephalopathy with de novo mutations in two novel genes: Chromodomain Helicase DNA Binding Protein 2 (CHD2) and Synaptic Ras GTPase Activating Protein 1 (SYNGAP1) (27). A second study sequenced 35 known or potential candidate genes in 53 epileptic encephalopathy patients (59), resulting in the identification of a number of causal de novo mutations in previously known genes.
De Novo Mutations in Epilepsy
When looking at epilepsy more generally, there has still been a strong focus in the community on the identification of de novo mutations. This has resulted in an increasing number of candidate genes. In fact, in a Pubmed search (June 20, 2013) for the term “De novo mutations + seizure” returned 244 primary papers highlighting a gene or structural variant linked to clinical manifestations of seizure. Within these papers, 65 unique genes have been identified, as well as 91 structural variants. Several genes were reported in multiple papers, including SCN1A (35), CDKL5 (11), PCDH19 (9), and KCNQ2 (7). Further classification of these genes revealed that 11 are known epilepsy genes (12), 11 are associated with epilepsy in the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), 16 are included on an epilepsy NGS gene panel (28), eight are listed with an association to a known “condition with seizures” on Genetics Home Reference (http://ghr.nlm.nih.gov), two genes harbor de novo mutations in patients with Autism Spectrum Disorder (55,56,60,61), one is a mouse seizure susceptibility gene (62), four are known ion channel genes (63), and 12 do not fall into any of these categories.
PHARMACOGENOMICS
Genetic variation can not only confer disease risk or protection but may also influence how a patient responds to medications. The study of the role of genetics in pharmacologic response, or pharmacogenetics, is particularly relevant in epilepsy disorders, since nearly all epilepsy patients undergo drug therapy for some period of time, and an estimated 30% of patients become drug resistant and never achieve seizure freedom despite treatment with all available antiepileptic medications.
Several genetically based hypotheses exist for why some patients fail medications that can control seizures in other patients with the same diagnosis. One hypothesis, the pharmacokinetic hypothesis, is that genetic variation causes differences in the absorption, metabolism, or distribution of antiepileptic drugs in the brain (64). In this model, a drug-responsive patient receiving a medication will get adequate concentrations of medication to the brain, whereas a drug-resistant patient will be unable to achieve sufficient medication levels in the brain to achieve seizure control. To date, the only consistently replicated evidence supporting this hypothesis is the association of variants in genes encoding a drug-metabolizing enzyme, CYP2C9, with phenytoin (a common antiepileptic drug) metabolism (65–67). Given the relatively minor consequences of the variation on the antiseizure response of phenytoin, the clinical utility of using CYP2C9 genotype to predict phenytoin dosage requirements is unclear. Despite this uncertain clinical relevance, dosing adjustments have been published recommending a 25% reduction in maintenance dose for the CYP2C9 *1/*2 and *1/*3 genotypes; a 50% reduction in maintenance dose for the CYP2C9 *2/*2, *2/*3, and *3/*3 genotypes; and cautious monitoring for adverse drug reactions associated with phenytoin toxicity (sedation, ataxia, nystagmus, dysarthria) (68). An alternative hypothesis is the pharmacodynamic hypothesis, where the gene encoding a drug target is mutated, and this change prevents the medication from being able to effectively modulate the intended pathway (69). There are no consistent reports of mutations in genes encoding drug targets or their modulators associating with response to antiepileptic dosing. It should be noted, however, that pharmacogenetic studies of antiepileptic medications performed to date have primarily evaluated the role of common variants (MAF >5% in the population) and are often statistically underpowered to robustly detect genetic associations. Larger studies considering less common variation are needed to better explore the aforementioned hypotheses.
In addition to there being little evidence to support either the pharmacokinetic or pharmacodynamic genetic hypotheses, they also fail to explain why a large number of drug-resistant patients fail to respond to multiple medications that are substrates for many different transporters at the blood–brain barrier and have differing modes of pharmacologic action. That is, under both of these hypotheses, many universally drug-resistant patients would have to have multiple mutations in genes encoding drug transporters governing drug disruption, metabolic pathways mediating metabolism, and/or pharmacologic targets. While it is possible that some patients may have acquired diffuse nongenetic changes due to seizures that are responsible for multidrug resistance, some patients present at the onset with drug-resistant seizures, and, despite aggressive early interventions, fail to respond. Recently, it has been proposed that drug-resistant epilepsy may reflect an intrinsically more severe form and that seizure control is therefore more difficult from the onset (70). One interesting additional possibility stemming from this hypothesis is that certain forms of epilepsy are not necessarily intrinsically more severe, but rather they arise from pathophysiologic changes that are not correctable with current pharmacologic agents. Therefore, what appears to be more severe epilepsy may in fact be a specific subtype of epilepsy with a currently unknown biologic etiology. Additional research is needed to reveal the neurobiological and genetic aspects of drug resistance, particularly as it pertains to the interplay of drug resistance and underlying pathophysiology.
Genetic variation can also dictate if a patient will experience severe adverse drug reaction to a medication. For antiepileptics, two clear genetic associations of HLA (human leukocyte antigen) alleles with cutaneous hypersensitivity reactions have been identified. First, in 2004, Chung et al. first reported a strong association between the presence of the HLA-B*15:02 allele and carbamazepine-induced Stevens-Johnson syndrome, a severe hypersensitivity reaction, in patients of Han Chinese descent. In this study, the HLA–B*1502 allele was present in 100% (44/44) of patients with carbamazepine-induced Stevens-Johnson syndrome, while only 3% (3/101) of patients on carbamazepine with the HLA–B*1502 allele had no reaction (71), indicating that the presence of this allele is highly predictive of this severe reaction to carbamazepine. These results were later replicated in the Han Chinese population and in other Asian populations, and were expanded to include toxic epidermal necrolysis, another severe cutaneous hypersensitivity reaction (72). Based on these findings and the frequency of the HLA–B*1502 alleles in Asian populations, the Food and Drug Administration currently recommends that all high-risk populations, including individuals of Han Chinese descent, and individuals from Vietnam, Cambodia, the Reunion Islands, Thailand, India, Malaysia, and Hong Kong, be screened for the presence of the HLA-B*1502 allele prior to initiating carbamazepine drug therapy. Limited evidence suggests that the HLA-B*1502 allele may also increase the risk of phenytoin-induced severe hypersensitivity reactions (73), which has led to the inclusion of a statement of caution in the drug label for phenytoin.
In addition to the risk of hypersensitivity reactions associated with the HLA-B*1502 allele, two recent GWA studies in patients of European and Japanese ancestry showed an association of an allele of the gene encoding the human leukocyte antigen A (HLA-A*3101) was associated with a range of carbamazepine-induced hypersensitivity reactions, including maculopapular exanthema, hypersensitivity syndrome, and the more severe Stevens-Johnson syndrome and toxic epidermal necrolysis (74,75). This association was not as strong as that seen for HLA-B*1502, with only moderately increased risk for hypersensitivity reactions (74,75). Unlike HLA-B*1502, HLA-A*3101 does not increase the risk of hypersensitivity to other aromatic antiepileptic medications, including lamotrigine and phenytoin (76). There are no current recommendations on genotyping HLA-A prior to carbamazepine treatment.
CLINICAL GENETIC TESTING
There are several varieties of genetic testing, including biochemical assays, cytogenetics, Sanger sequencing (typically used for single gene testing), and NGS (used for gene panel sequencing or whole-exome sequencing). Genetic testing can determine a genetic diagnosis, which, in turn, helps guide patient care and counseling about prognosis and reproductive choices for the parents. In addition to diagnostic testing, predictive testing may be valuable for patients with a family history of epilepsy. In the United States, all genetic testing that will be used for medical management or intervention for a patient must be performed in a Clinical Laboratory Improvement Amendments (CLIA)–certified laboratory. Access to a genetic counselor should be provided to help the patient and their family understand the results and their implications.
It is critical to be aware of the current speed of genetic discovery because this dictates the growth in the field of genetic testing, with new information emerging practically every day. In epilepsy genetic testing, there are guidelines established for the clinical utility of testing the most commonly mutated genes (12,44). For example, screening of SCN1A in Dravet syndrome or CDKL5 in infantile spasms are useful screens because mutations (typically de novo) explain 70% to 80% and 10% to 17% of cases, respectively (11). In patients experiencing seizures with cortical malformation, one of the most common diagnoses is periventricular nodular heterotopia (PNH). The most common cause of familial PNH and nearly 25% of the nonfamilial forms are caused by mutations in the FLNA gene; again making this a logical first diagnostic screen (43). For a small proportion of genes, knowing the genetic etiology can guide treatment, as is the case for SLC2A1 mutation–positive patients (“GLUT1 deficiency syndrome”) where the ketogenic diet is the gold standard of treatment (77).
Chromosomal microarray analysis and karyotyping should be used to detect structural abnormalities; this is particularly critical for patients with seizures who exhibit mild or moderate intellectual disability and/or dysmorphic features. In this case, fragile X testing and biochemical testing of amino acid levels may also be useful (43). CNV screening appears to be critical in two main groups: epileptic encephalopathy (CNVs explain ~4% of cases) (43) and GGE with intellectual disability (CNVs explain ~10% of cases) (78).
In patients where the first genetic screen is less obvious, or if screening of multiple genes has failed to identify the disease-causing variant, ordering a comprehensive panel of genes is a very practical approach. For many diseases, including epilepsy, gene panels are offered for genetic testing; gene panels target a set of genes that are associated with a given disorder. Sequencing of these genes is usually achieved by NGS of the coding exons and the flanking intronic boundaries. The set of genes included may vary by the company performing the CLIA-certified sequencing. For example, GeneDx offers a number of different epilepsy gene panels. The most comprehensive panel includes 71 genes, but smaller panels are available that target infantile of childhood-onset seizures. The Courtagen epiSEEK gene panel includes over 300 genes. Current lists of all available genetic tests and testing facilities are available online and updated regularly (http://www.genetests.org and http://www.orpha.net).
From a clinical perspective, it is practical to order a phenotypically relevant gene panel than to order exome sequencing. One reason for this is that variants identified in genes selected for a gene panel will be more interpretable based on prior knowledge; in contrast, opening interpretation up to the exome may generate a long list of variants of “unknown clinical significance.” In addition, this avoids issues associated with “incidental genetic findings,” which may arise when interrogating the exome. However, if the patient has a very unique phenotype, exome-sequencing may be a more desirable approach than a gene panel. Finally, it is usually helpful to also have parental DNA available for testing to determine the inheritance (ie, de novo status) and thus likely pathogenicity of any identified variants.




