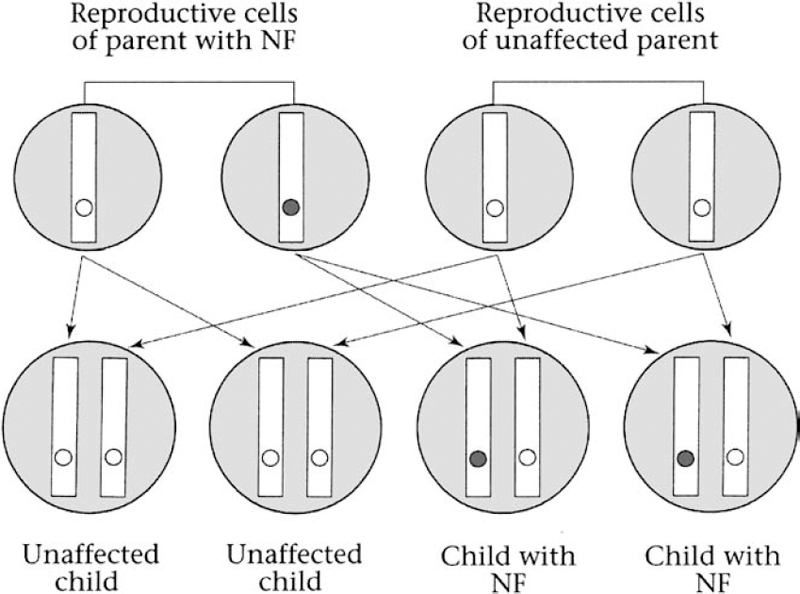
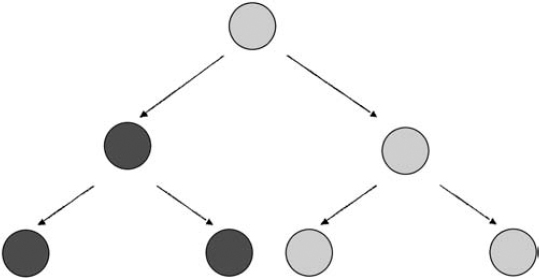
4 Molecular genetics explains how a mutation may cause a disorder. Clinical genetics takes a somewhat different perspective: how the mutation is inherited, how likely it is that the mutation will cause a disorder, and what medical and ethical decisions might face the patient and family. Some of the major components of clinical genetics, as it relates to neurofibromatosis 1 (NF1) and neurofibromatosis 2 (NF2), are explored in this chapter. (Schwannomatosis appears to have a hereditary pattern that is different from that of NF1 and NF2, and the biological mechanism that causes it is not yet known. For this reason, the issues of cause and genetic counseling are discussed in Chapter 12.) NF1 and NF2 both originate because of a change in a gene (i.e., the NF1 or NF2 gene). If a child with neurofibromatosis (NF) has a parent with NF, the condition has been inherited. If a child is diagnosed with the disorder but neither parent is affected, the condition probably developed because of a spontaneous mutation in an egg or sperm cell. At this time, there is no way to prevent NF1 or NF2. Genes mutate constantly in the body, and these genes have an unusually high rate of mutation, probably because both are relatively large and therefore there are more opportunities for error. Many parents feel guilty when a child is diagnosed with NF, especially if there is no family history of the disorder. However, nothing a parent does (or doesn’t do) can cause or prevent NF1 or NF2. These disorders are not caused by exposure to toxins, radiation exposure, or alcohol consumption. NF1 and NF2 are autosomal dominant conditions, which means that there is a 50/50 chance of passing the disorder on every time a person who has the disorder has a child. Genetic tests for NF1 and NF2 are now becoming available for routine clinical testing. These tests can be expensive, however, due to the complexity of finding the sometimes subtle mutations in the large NF1 and NF2 genes. Currently gene therapy does not exist for NF1 and NF2. Scientists still have much work to do in understanding the basic disease process in both disorders before they will be able to develop interventions. In the meantime, many options exist for management. Once a person is born with the genetic mutation responsible for NF1 or NF2, there is a 50/50 chance of passing the condition on whenever that person has a child. To understand how offspring inherit these disorders, consider the principles of dominant inheritance. Everyone has two copies of each gene. Most cells in the body hold both copies, contained in 46 chromosomes. The exceptions are egg and sperm cells, which contain only 23 chromosomes, and thus only one copy of each gene. When egg and sperm unite, 46 newly paired chromosomes result. Thus, there are four possible genetic combinations every time egg and sperm unite (Fig. 4–1). This explains why, for people who have NF, there is a 50% chance in every pregnancy for the child to have neurofibromatosis. Obviously, the decision about whether or not to have a child can be difficult for someone with NF1 or NF2. The risk of passing the disorder on is akin to a coin toss, and yet the ramifications are much more serious. This is a decision that should be made carefully, and after consultations with the partner, other loved ones, a genetic counselor, and, for some people, a spiritual advisor. Both the NF1 and NF2 genes are highly penetrant, which means that if they are inherited they will cause manifestations. The real question is which manifestations and how severe those manifestations will be. Figure 4–1 Two illustrations on left: A child will not inherit neurofibromatosis if two unaffected reproductive cells unite. Two illustrations on right: A child will inherit the disorder if an affected cell is united with an unaffected cell. (Courtesy of Harriet Greenfield, with permission.) When NF1 or NF2 appears in a child born to parents who do not have the disorder, the typical reaction is: How could this happen? There are two possible answers. One is that a parent may in fact have NF1 or NF2, but the manifestations may be so mild that he or she does not realize it. The other explanation is that a mutation developed in either the sperm or egg cell that were joined at fertilization and eventually developed into the affected child. Like other cells in the body, sperm and egg cells constantly divide as they develop and mature. Egg cells divide while a female fetus is still developing in the womb; consequently a woman is born with all the eggs she will ever have. Sperm cells continue dividing through a man’s lifetime. As discussed in Chapter 3, any time a cell divides there is a chance that a gene may mutate. Each time a cell divides, it first makes copies of thousands of genes, and there is always the chance that a copying error will occur sometime during the process. The genes that cause NF1 and NF2 are relatively large and therefore exceptionally prone to such copying errors. In fact, it is likely that most people have some egg or sperm cells that contain a mutated version of the NF1 or NF2 genes. When one egg or sperm cell containing a mutated NF1 or NF2 gene is involved in conception, the child who results will have the disorder. When a sporadic case of NF1 or NF2 occurs, parents often wonder whether other children in the family will also have NF. In most cases, they will not, unless, by chance, there is another conception involving an egg or sperm with the requisite genetic mutation. It is unlikely (though not impossible) that parents who do not have NF will give birth to another child with NF1 or NF2. The child with NF, however, will have a 50/50 chance of giving birth to offspring with NF1 or NF2. Although most sporadic cases of NF1 and NF2 occur when a chance mutation develops in either the sperm or egg cell that unite at conception to form a zygote, the resulting single fertilized cell, some occur because of a mutation that takes place later in prenatal development. Such postzygotic mutations develop into mosaic or segmental forms of neurofibromatosis. This person is born with a mixture of cells, some with two functioning copies of the NF1 or NF2 gene, and some with mutated copies (Fig. 4–2). If the postzygotic mutation occurs very early in embryonic development, before cells have begun to differentiate (take on specialized functions), most cells in the body will carry the mutation, and so the manifestations will be clinically indistinguishable from other cases of NF1 and NF2. If the postzygotic mutation occurs later in prenatal development, after cells have differentiated into different tissues and organs, manifestations may be milder or may result in only some parts of the body. An example of this is a case of NF2 where tumors develop in only one ear (known as unilateral vestibular schwannoma), and involve only one region of the body.1 Another example, more controversial, is that of mosaic NF1, in which neurofibromas and pigmentary changes such as café-au-lait spots develop in only one area, such as one quadrant of the body. Figure 4–2 Mosaic forms of neurofibromatosis develop when a genetic mutation occurs during embryonic development. The child is born with a mixture of cells—some containing the NF mutation and some without. The condition is transmitted to offspring only when egg or sperm cells carry the mutation. (From Korf BR. Human Genetics: A Problem-Based Approach. Oxford: Blackwell Science; 2000:60, with permission.) The issue of mosaicism is probably of greater interest to genetic researchers than to patients and physicians. The condition can be confirmed only through genetic analysis of cells from different parts of the body. Clinically speaking, people with mosaic forms of NF1 and NF2 tend to have less severe manifestations, and they may be less likely to pass the disorder on to children. However, given the small likelihood that a case of spontaneous NF1 or NF2 is mosaic, and the difficulty in determining this, genetic counseling remains the same whether a person has the mosaic form or not. Genotype refers to a person’s unique collection of genes, whereas phenotype refers to the physical characteristics that result when instructions from those genes are decoded. Both the NF1 and NF2 genes are prone to multiple types of mutations that affect manifestations of these disorders. There are no clear genotype-phenotype correlations in NF1; so far we cannot say which type of mutation can be linked to what type of clinical manifestation. The exceptions to this general rule are mutations that involve large or entire deletions of the gene. People with NF1 who have a large deletion mutation in the gene tend to develop neurofibromas earlier than expected, more significant developmental delays, and other physical anomalies.2 In most cases, however, it is so far impossible to predict how mild or severe the disorder will be based on an analysis of the gene mutation or, in inherited cases, on the basis of other family members’ manifestations. On the other hand, there appear to be strong genotype/phenotype correlations in NF2. Members of the same family usually display the same manifestations and severity. It is possible, therefore, to predict with confidence how severe the manifestations of NF2 will be in people with the familial form of the disorder. Genetic tests used in research may seek to characterize the type of mutation, determine whether it is inherited or acquired, and discover how the mutation affects the gene’s protein product and an individual’s physical characteristics, or phenotype. A variety of genetic tests exist for both NF1 and NF2, and in the research setting they have yielded many insights into the functioning of the NF1 and NF2 genes. (To learn more about how to participate in such research studies, see Chapter 15.) Clinical genetic testing uses many of the same methods, but for a different reason: to determine whether someone has a gene mutation associated with a specific disorder. Clinical testing may be used to confirm a suspected diagnosis, or to establish the type of mutation as a prelude to offering prenatal diagnosis to a couple where one partner has the disorder. Because of the complexity of the NF1 and NF2 genes, genetic testing does not detect all possible mutations. The area of clinical genetic testing is changing rapidly, so consult your physician or a genetic counselor for up-to-date information. Various types of genetic tests are available. Direct DNA testing examines the gene sequence segment by segment, and it provides the most specific information regarding the nature of a genetic mutation. This test predicts nothing about disorder severity. This method can be time consuming and expensive in large genes, such as NF1 and NF2. A protein truncation test determines whether a gene’s protein product (such as neurofibromin in NF1 or merlin in NF2) is altered in such a way that no protein or not enough protein is produced, which provides an indirect indication of whether a gene has mutated. The test is not specific enough to determine the particular mutation, however, unless it is followed by genetic sequencing. Fluorescence in situ hybridization (FISH) testing can determine whether the entire gene is deleted, but it is of value only to the small number of people who may have the “large deletion” NF1 phenotype. Linkage analysis takes DNA samples from affected and unaffected family members to track the manner by which the mutation is passed through the family. It is of value only to people who have the familial form of NF1. This type of testing requires not only that affected family members from three generations be available and willing to be tested, but also that the diagnosis be accurate in all those tested, and that genetic relationships be clear. The decision about whether to obtain a genetic test can be difficult. Any type of genetic test has risks as well as benefits; thus the decision should be made only after weighing all the factors. Some guidance about the issues is provided in the section on genetic counseling. The chameleon nature of NF1 and the length of the NF1 gene make genetic testing complicated. Such testing is now clinically available and can be used to confirm whether an NF1 mutation is present in 95% of people tested. As mentioned earlier in this chapter, however, so far little is known about specific genotype-phenotype correlations in NF1, which means that even if a genetic mutation is detected in someone, it does not help a physician to predict what manifestations a person will experience. Moreover, genetic testing provides no information as yet that would serve to improve therapy in people with NF1. For these reasons, most cases of NF1 can be diagnosed and managed by a physician using the expert consensus clinical criteria (see Chapter 5). There are two types of situations in which clinical genetic testing for the NF1 gene mutation might be useful: ♦ Confirming a Diagnosis In some unusual cases, a diagnosis of NF1 cannot be confirmed through any method other than a genetic test. The situation usually involves a child who develops multiple café-au-lait spots and is suspected of having NF1, yet is born with no family history of the disorder. In most situations, the best strategy is watchful waiting and conducting further clinical tests at the appropriate times (such as having the child examined by an ophthalmologist to detect the presence of Lisch nodules, which can be used to confirm a diagnosis). Because several years may pass before new signs of the disorder are discernible, parents sometimes become so anxious that it is difficult for them to wait. In this situation, a clinical genetic test could help to ease their anxiety by providing a definitive answer one way or the other. Another situation in which genetic testing might be helpful is when one of the parents has NF1 and a child displays minimal signs of having the disorder, such as four café-au-lait spots. In this case, testing may help determine whether the child has NF1. There are also times when a child or adult presents with unusual features of NF1, such as delayed onset of puberty or epilepsy, which may represent a variant form of the disorder. In this case, testing may help to clarify what is causing the manifestations. ♦ Prenatal Testing It is possible to test a fetus for an NF1 gene mutation if a parent has NF1 and is concerned about passing the disorder on to offspring. Samples of fetal DNA are obtained using either amniocentesis (a sample of the amniotic fluid surrounding the fetus, to detect any chromosomal abnormalities) or chorionic villus sampling (analysis of a tissue sample taken from the placenta to detect any genetic abnormalities). Direct analysis of fetal DNA is one possibility.3 Linkage analysis provides another option for testing in people with the inherited form of NF1, as long as enough affected family members are available and willing to be tested.4 As with adults, however, detecting a mutation does not predict how severely or mildly offspring will be affected; therefore, a genetic test is of no value in terms of prognosis. If parents request such testing to make a decision about whether or not to continue the pregnancy, then genetic counseling should be available so that they can make an informed decision. Genetic tests for NF1 are now available on a clinical basis. Because of the large size of the NF1 gene and the wide variety of possible mutations, it is difficult and costly to identify a particular mutation. A limited number of hospitals, universities, and diagnostic laboratories in the world provide such testing, and then only at the request of a physician (not a patient) and usually only for certain well-defined situations described above. In general, it is better to use clinical criteria for diagnosis and to monitor manifestations as they develop. Because there is a strong degree of genotypic-phenotypic correlation in inherited NF2, genetic testing is sometimes useful in people with this disorder. Genetic tests are used most often to identify children who are at risk of inheriting NF2 because they are born into families with a history of the disorder. Once such a child is confirmed to have an NF2 mutation, he or she can be monitored periodically for development of manifestations so that treatment can be administered at appropriate times. These tests have some limitations, however. Direct DNA tests that are currently available find mutations in ~65% of people tested.5,6 Therefore, they are not completely accurate. As a result, even a child who tests negative for an NF2 mutation should continue to be monitored until early adulthood to ensure that manifestations do not develop. Another caution is that the tests are not yet sensitive enough to predict phenotype. Linkage analysis is more accurate, providing that other family members agree to participate. Although still difficult to access, genetic testing for NF2 is emerging. Clinical testing is available at a handful of sites throughout the world, including two in the United States. Only physicians may request such testing for a patient. As was demonstrated in the previous chapter, the pathogenesis of NF1 and NF2 is quite complex and difficult for most patients and families to understand. For that reason, many people who have neurofibromatosis or have a child with the disorder will benefit from a genetic counseling session. Although specifics vary, the goal of genetic counseling is to translate complicated medical information in a way that a layperson can understand. The genetic counselor will then help the person with a genetic disorder (or the parents) understand the issues involved in various decisions, such as choosing among treatments or deciding whether to have a child. What a genetic counselor does not do is actually make those decisions; his or her role is to explain the risks and benefits and provide estimates of how likely it is that complications might occur. Often the first step in a genetic counseling session is to confirm a diagnosis. To do so, the genetic counselor may review individual medical records, ask questions about family medical history, and review results from medical tests. He or she may also conduct a physical exam. The next step usually involves a discussion of what manifestations the disorder causes, how those manifestations develop, and what options exist for treatment. The counselor should also discuss how the disorder can be passed on to offspring. By the end of the session, the person seeking counseling should understand the genetic disorder better and feel more empowered to make decisions. Receiving a diagnosis of NF1 or NF2 can be upsetting, especially if there is no family history of the condition and it is discovered incidentally. As a result, it’s normal to feel confused, anxious, scared, and depressed. Patients should expect, and health care providers should be prepared, to spend time addressing emotional as well as medical concerns. Patient education brochures and other educational materials are helpful, as are referrals to support groups and patient advocacy organizations. Nancy B.: “My twin sister and I look a lot alike, but we didn’t know if we were identical. We actually found out through a clinical trial. This was probably 15 or 20 years ago, when I was 30, and we had genetic testing done. The test involves doing more and more refined DNA probes until either a difference is found or they give up. In our case, they did give up and we were told that there was a 99.9% chance that we are identical. The interesting thing is that even though my sister and I are identical, we have some of the same manifestations and some different ones. You’d like to think that there is a genotype-phenotype correlation, but that is not the case in NF1. “I didn’t get married until I was 37. I had already decided that I would not have any children, so that was of course something we discussed before getting married. Because NF is a dominant trait, there is a 50% chance that each of our children might have NF. More importantly, there is not a way to predict severity. I knew I couldn’t live with myself if I had a child who was severely affected. I’d feel guilty about causing emotional and physical pain to an innocent child. So that was always a concern when I was dating. When I got serious about the man who is now my husband, I knew I had to get this out in the open. Everyone should be able to have a child without worries like these. Fortunately, not having children wasn’t an issue for him, and we have been happily married for 13 years. Still, I often think about what it would be like to have children.” Kellie C: “I knew there was a 50/50 chance that each of my children could inherit NF1. We basically put our faith in God. We came to the conclusion that no one is guaranteed a perfect pregnancy or a healthy child. We have two kids. David is 4 1/2 years old and Danielle is 19 months old. David definitely does not have NF1. Danielle has three or four café-au-lait spots, so we’re watching her. Our primary care physician delivered both my children. And when I was pregnant, she said, ‘Worse things can happen than NF1.’ I think people need to remember that most cases of NF1 are mild. The chances are low that NF1 will be severe. I think you need to keep it in perspective.” Dolores G.: “My daughter Susan was diagnosed with NF1 in the early 1970s. The doctors did try to sit down and explain what NF was, but the information was all wrong. At that time the consensus was that NF had to be passed on by one of the parents. There was no talk of spontaneous mutations. My husband and I were both examined at the hospital. I have a lot of freckles, so therefore I was diagnosed with NF. As it turns out, I didn’t have it. I was ‘de-diagnosed’ later, as I like to say. “But at the time, they decided I had NF1 also, because of the freckles. We sat down with the head of the hospital. He was a personal friend and ex-boss of mine. And we talked about this. My husband and I both asked him, what do you think that we should do? We’d like to have more children. Even before we got married we said we would adopt as well as have biological children. But then I got pregnant with Susan, so our plans to adopt were postponed. He said that if he was in our position, since we already had two daughters, that he would probably not have other kids because the chance of having another child with NF1 was so high. And you only needed one parent to pass NF on.” Adam G.: “I know there’s a 50% chance that I could pass this on to my children. I am concerned about it. One of my biggest faults is that I always think too far in the future. I consciously try not to think about this whole issue yet. It is a concern, but I try not to let it get to me right now. My mom says that by the time it’s an issue, there may be a cure for NF2.” Martha L.: “My husband and I don’t have children. As much as I love life, and kids, I decided I didn’t want to put anyone through the life I have had to lead. My husband completely understood and knew before we got married how I felt about it. We don’t have a history of NF in my family, so my parents didn’t know that any of their children would have NF. I am the only one out of three that has it. Tamra M.: “I’ve thought about children a lot. I talked it over with one of my teachers who was close to me. We were talking one day and she asked me whether I would have children, and I wasn’t sure. A year later, I did a report on orphans and homeless children. I found out how many orphans there were. So I thought, there’s the solution! I’m thinking I’ll adopt children because there are so many who need homes.” Angela W.: “I knew I wanted to have children more than anything. We thought we’d wait a year or two after we got married. God kind of made the decision for us. We decided it was a blessing and God’s will, so we were going to go ahead with it and let nature and fate decide what happens. “We did talk with a genetic counselor when I was 5 or 6 months pregnant. It helped my husband more than it helped me. I knew a lot of the information already, but it was hard to explain to someone else. The counseling session helped my husband understand how NF1 can be inherited and that the pregnancy might worsen some of my symptoms. “I think if you have NF1 and you’re thinking of having kids, you need to be willing to accept whatever problems there might be. Know what you’re getting into. So far my baby looks healthy and I’m happy with that. But if Davin has complications down the road, I have no regrets. I’m willing to accept whatever comes.”
Genetics and Genetic Counseling in Neurofibromatosis 1 and Neurofibromatosis 2
♦ Common Questions About the Genetics of Neurofibromatosis 1 and Neurofibromatosis 2
What causes Neurofibromatosis 1 and Neurofibromatosis 2?
Can These Disorders Be Prevented?
What Is the Risk that a Parent Will Pass Neurofibromatosis on to the Child?
Is Genetic Testing Available?
Is Genetic Therapy Available?
♦ Principles of Autosomal Dominant Transmission
How Familial NF1 and NF2 Are Inherited
Spontaneous Cases of Neurofibromatosis 1 and Neurofibromatosis 2
Mosaicism in Neurofibromatosis 1 and Neurofibromatosis 2
♦ Genotype/Phenotype Correlations in NF1 and NF2
♦ Genetic Testing for Neurofibromatosis 1 and Neurofibromatosis 2
Neurofibromatosis 1 Testing Issues
Clinical Indications for Testing
Availability
Neurofibromatosis 2 Testing Issues
Clinical Indications for Testing
Availability
♦ Genetic Counseling
♦ The Personal Perspective
References
< div class='tao-gold-member'>
Genetics and Genetic Counseling in Neurofibromatosis 1 and Neurofibromatosis 2
Only gold members can continue reading. Log In or Register to continue






Full access? Get Clinical Tree


