Proposed inheritance
Number of families
Affected members
Reference study
Autosomal dominant with reduced penetrance
21 families
Parent–child, siblings, avuncular pairsa, cousins
Milhorat et al. (1999)
23 families
Parent–child, siblings, avuncular pairs, cousins
Boyles et al. (2006)
1 family
Two brothers
Robenek et al. (2006)
31 families
Parent–child, siblings, avuncular pairs, cousins
Speer et al. (2000)
1 family
2 monozygotic twins and first-degree relatives
Stovner et al. (1992)
1 family
3 generations
Coria et al. (1983)
1 family
3 affected members
Giménez-Roldán et al. (1978)
Autosomal recessive
21 families
Siblings, avuncular pairs, cousins
Milhorat et al. (1999)
Multifactorial
3 families
Parent–child, monozygotic twins, avuncular pairs and cousins
Szewka et al. (2006)
Undetermined
3 families
2 mother–daughter pairs and 1 father–daughter pair
Schanker et al. (2011)
15 families
15 surgically treated cases with positive family history including 3 pairs of affected siblings
Tubbs et al. (2011)
1 family
4 generations
Tubbs et al. (2004a)
1 family
3 sisters
Weisfeld-Adams et al. (2007)
31 families
Parent–child, siblings, avuncular pairs, cousins
Speer et al. (2000)
1 family
2 sisters
Mavinkurve et al. (2005)
1 family
Monozygotic twin sisters and the daughter of one sister
Atkinson et al. (1998)
1 family
2 siblings
Stovner and Sjaastad (1995)
1 family
2 siblings
Herman et al. (1990)
Families with Chiari type I malformation showed both vertical (mother-to-child) and male-to-male (father-to-son) transmission1 consistent with an autosomal dominant mode of inheritance . Since the disease frequency in these affected families is less than what would be expected from pure Mendelian inheritance, Chiari type I malformation is thought to be incompletely penetrant.2 Other pedigree studies, however, have implicated autosomal recessive mode of inheritance for Chiari type I malformation (Table 5.1). Most likely, the pattern of inheritance is oligogenic, i.e. determined by the cumulative effect of variants in several genes, albeit with variable penetrance.
A few cases have been reported of familial syringomyelia without an associated Chiari type I malformation (Robenek et al. 2006; Koç et al. 2007), although another study found no cases of familial syringomyelia in the absence of Chiari type I malformation, in a cohort of over 150 families (Speer et al. 2003). It may be that cases of ‘isolated’ familial syringomyelia have a volumetrically small posterior fossa without overt tonsillar herniation (Mavinkurve et al. 2005).
5.4.2 Twin Studies
Classical twin studies compare the occurrence of the same trait or disease in monozygotic and dizygotic twins . Monozygotic twins develop from a single fertilised egg and therefore have identical genetic material. Dizygotic twins derive from two eggs that were fertilised independently from two different sperm cells at the same time. These twins, like any other siblings, share 50 % of their genes.
Comparing the concordance 3 of monozygotic twins for a trait or disease with that of dizygotic twins provides an estimate of the extent to which genetic variation contributes to that trait or disease. A higher concordance in monozygotic as opposed to dizygotic twins indicates a genetic contribution to the trait under study (Boomsma et al. 2002). Several twin studies of Chiari type I malformation have reported an almost 100 % concordance in monozygotic twins (Stovner et al. 1992; Iwasaki et al. 2000; Szewka et al. 2006; Miller et al. 2008; Tubbs et al. 2008a; Solth et al. 2010). Only five studies were examined for an associated syringomyelia, and three sets of twins were found to be discordant for this phenotype (Stovner et al. 1992; Iwasaki et al. 2000; Tubbs et al. 2008a), whilst two other sets were concordant for the absence of syringomyelia (Miller et al. 2008; Solth et al. 2010). Another report described syringomyelia in monozygotic twin brothers who were discordant for Chiari type I malformation (Tubbs et al. 2004b). A unique report of a monozygotic triplets described differing degrees of tonsillar descent; one triplet was affected by Chiari type I malformation and syringomyelia, whilst the other two asymptomatic siblings had tonsillar descent of 4 and 2.5 mm, respectively (Cavender and Schmidt 1995). A study of three pairs of dizygotic twins revealed that one pair of sisters was concordant for Chiari type I malformation with syringomyelia. A second pair of sisters had Chiari type I malformation but only one of them had syringomyelia. In a third pair, one sister had Chiari type I malformation with syringomyelia while the female co-twin had neither (Speer et al. 2003). Collectively, these studies indicate a higher concordance of Chiari type I malformation between monozygotic than dizygotic twins, further supporting a genetic basis for Chiari type I malformation. Clearly, additional, larger twin studies are needed to confirm these findings and to investigate further twin concordance for syringomyelia associated with Chiari type I malformation.
5.4.3 Association with Known Genetic Syndromes
Co-segregation 4 of one condition, with one or more other known genetic conditions, suggests a genetic basis for the first condition. The assumption is that a common genetic defect is responsible for the various abnormal phenotypes within the complete syndrome. Chiari type I has been associated with several known genetic disorders or syndromes (Table 5.2). The majority of these disorders affect bone structures, for example, achondroplasia and Crouzon syndrome, or pathways involved in axial mesodermal growth and differentiation, for example, Williams syndrome and Shprintzen–Goldberg syndrome . The causative genes have been identified for some of these conditions and are mainly regulators of signalling pathways or transcription factors.5 A few are implicated in essential cellular functions, such as chromatin methylation and proteolysis. DNA methylation plays an important role in regulation of gene expression during development and differentiation (Qureshi and Mehler 2011). Proteolysis is the process by which proteins are hydrolysed into small peptides and removed or cleaved for cell signalling (Maupin-Furlow 2011). The genes are hypothesised to have pleiotropic effects 6 on the manifestation of cerebellar tonsil herniation, occipital hypoplasia , syringomyelia and other phenotypes.
Table 5.2
Summary of genetic diseases and syndromes that can be associated with Chiari type I malformation
Syndrome (phenotype MIM#a) | Locus | Gene | Gene function | References |
|---|---|---|---|---|
Signalling transducers | ||||
Achondroplasia (MIM# 100800) | 4p16.3 | FGFR3 (fibroblast growth factor receptor 3) | Transmembrane growth factor receptor that mediates FGF signalling during development | |
Costello syndrome (MIM# 218040) | 11p15.5 | HRAS (v-Ha-ras Harvey rat sarcoma viral oncogene homologue) | Member of the Ras oncogene family that functions in signal transduction pathways | |
Crouzon syndrome (MIM# 123500) | 10q26.13 | FGFR2 (fibroblast growth factor receptor-2) | Transmembrane growth factor receptor that mediates FGF signalling during development | |
Apert’s syndrome (MIM#101200) | ||||
Hajdu–Cheney syndrome (MIM# 102500) | 1p12-p11 | NOTCH2 (Notch gene homologue 2) | Notch type 1 transmembrane protein that plays a role in bone metabolism | |
Klippel–Feil syndrome (MIM# 118100) | 8q22.1 | GDF6 (growth/differentiation factor 6) | Bone morphogenetic protein that regulates the formation of skeletal joints in the limbs, skull and axial skeleton | |
Loeys–Dietz syndrome type 1 (MIM# 609192) | 9q22.33 | TGFBR1 (transforming growth factor, beta receptor 1) | Serine/threonine protein kinase that functions in TGF-beta signalling | Loeys et al. (2005) |
Neurofibromatosis type I (MIM# 162200) | 17q11.2 | NF1 (neurofibromin) | Negative regulator of the Ras signal transduction pathway | |
Noonan syndrome-1 (MIM# 163950) | 12q24.13, | PTPN11 (protein tyrosine phosphatase, non-receptor type 11) | Protein tyrosine kinase that plays a role in signalling via the RAS-mitogen activated protein kinase (MAPK) pathway | |
Paget’s disease of bone (MIM# 602080) | Genetically heterogeneous | PDB4 (Paget’s disease of bone 4) | PDB4: undetermined function | |
5q31 | SQSTM1 (sequestosome 1) | SQSTM1: binds ubiquitin and regulates activation of the nuclear factor kappa-B (NF-kB) signalling pathway | ||
5q35.3 | TNFRSF11A (tumour necrosis factor receptor super family, member 11a) | TNFRSF11A: member of the TNF-receptor super family, an essential mediator for osteoclast and lymph node development | ||
18q21.33 | ||||
Transcription factors | ||||
Chromosome 1p32-p31 deletion syndrome (MIM# 613735) | 1p32-p31 | NF1A (nuclear factor 1) | CAAT box transcription factor, plays a major role in development | Lu et al. (2007) |
Cleidocranial dysplasia (MIM#600211) | 6p21.1 | RNX2 (runt-related transcription factor 2) | Transcription factor, a key modulator of osteoblast differentiation | |
Combined pituitary hormone deficiency-4 (MIM#262700) | 1q25.2 | LHX4 (LIM-homeobox 4) | Transcription factor, functions during the development of the mammalian pituitary gland and the nervous system | |
Type II blepharophimosis–ptosis–epicanthus inversus syndrome (BPES) (MIM# #110100) | 3q22.3 | FOXL2 (Forkhead box L2) | Forkhead transcription factor important in ovarian development | |
Velocardiofacial syndrome (MIM#192430) | 1.5–3.0-Mb hemizygous deletion of 22q11.2 | Some cases are caused by mutations in TBX1 (T-box 1) | TBX1: transcription factor with a conserved DNA-binding domain, the T-box | |
Miscellaneous functions | ||||
Cystic fibrosis (MIM# 219700) | 7q31.2 | CFTR (cystic fibrosis transmembrane conductance regulator) | Member of the ATP-binding cassette (ABC) transporter super family, functions as a chloride channel | |
Hypophosphatemic rickets (MIM# 307800) | Xp22.11 | PHEX (phosphate-regulating endopeptidase homologue, X-linked) | Zinc-dependent metalloprotease, found in the cell-surface membrane of osteoblasts, osteocytes and odontoblasts | |
Idiopathic growth hormone deficiency (MIM# 173100) | 17q23.3 | GH1 (growth hormone) | Growth hormone | |
Kabuki syndrome (MIM# 147920) | 12q12-q14 | MLL2 (myeloid/lymphoid or mixed-lineage leukaemia 2) | Trithorax-group histone methyltransferase, important in the epigenetic control of active chromatin states | |
Miller–Dieker lissencephaly syndrome (MIM#247200) | 17p13.3 | PAFAH1B1 (platelet-activating factor acetylhydrolase, isoform Ib) | Inactivating enzyme for platelet-activating factor, important for neuronal migration | Nagamani et al. (2009) |
Shprintzen–Goldberg syndrome (MIM# 182212) | 15q21.1 | FBN1 (fibrillin 1) in some cases | Extracellular matrix glycoprotein that serves as a structural component of 10–12 nm calcium-binding microfibrils | |
Associated genes unknown | ||||
Macrocephaly–capillary malformation (MIM# 602501) | Unknown | Unknown | ||
Primary basilar impression (MIM#109500) | Unknown | Unknown | Bentley et al. (1975) | |
Chromosome 16p11.2 rearrangements | Unknown | Unknown | Schaaf et al. (2011) | |
William’s syndrome or chromosome 7q11.23 deletion syndrome (MIM#194050) | Hemizygous deletion of 1.5–1.8 Mb on chromosome 7q11.23 | Unknown | ||
Alternatively, Chiari type I malformation could be acquired secondarily in some of these diseases, for example, in cystic fibrosis, consequent upon constant Valsalva, from recurrent coughing or wheezing or as a result of metabolic and electrolyte imbalances (Patel et al. 2011).
Genomic deletions or duplications , on chromosome 7q and chromosome 16p , have been associated with Chiari type I malformation (Pober and Filiano 1995; Mercuri et al. 1997; Ferrero et al. 2007; Schaaf et al. 2011). These rearranged chromosomal regions contain a large number of biologically plausible candidate genes for Chiari type I, including TBX6 , on chromosome 16p , that encodes a transcription factor important in establishing mesodermal identity and which can have a role in the aetiology of congenital spinal anomalies (Schaaf et al. 2011). A more comprehensive and systematic research of these regions is needed to identify the underlying genetic lesions and to understand their pathogenic role in the development of Chiari type I malformation.
5.5 Molecular Studies of Chiari in Humans
While Chiari type I malformation has a tendency to aggregate in families, it is rarely segregating in a classical Mendelian fashion. It is believed to be a complex trait that could be either oligogenic or polygenic, i.e. resulting from a large number of genetic variants, each contributing small effects. One cannot exclude the possibility of unknown environmental or nongenetic influences that may interact with these predisposing genetic factors to modulate the incidence of the Chiari type I with or without syringomyelia phenotype.
Studies of alleles7 that influence other complex diseases could provide some indication of what might be taking place with Chiari type I malformation and syringomyelia. There is considerable heterogeneity both as regards the frequency and as regards the strength of effect of the alleles described to date (Fig. 5.1). At one end of the spectrum are high risk alleles, segregating in large families affected with Mendelian diseases. These can be identified easily by family-based linkage studies that aim at identifying a polymorphic genetic marker allele8 that segregates strongly with the disease phenotype in a family.9 At the other extreme, gene identification in complex traits remains a challenge. A number of common alleles have been found to be associated with common phenotypes, as predicted from the common disease/common variant hypothesis. These common variants are usually identified by genetic association studies that investigate the association between common genetic variation and disease in a large numbers of study subjects. This type of analysis requires a dense set of polymorphic markers that capture a substantial proportion of common variation across the genome (for genome-wide association studies or GWAS10) or across a set of biologically plausible candidate genes (for candidate gene association studies) (Frazer et al. 2009). Common variants seem to have modest effect sizes.11 Even when combined, their impact on overall population variance and predictive power12 is limited. For many traits, associated variants have explained only a small proportion of estimated heritability .13 A significant proportion of this undetermined heritability, known as ‘missing heritability’, may be attributable to variants that are of low frequency (<0.01 in frequency) with intermediate penetrance effects, which cannot be detected by conventional gene-discovery approaches mentioned above (Manolio et al. 2009). Recently, a role for rare de novo mutations is emerging in the genetic architecture of some of the complex traits, particularly those that decrease the reproductive fitness and incur a large degree of selection against the phenotype (Gillis and Rouleau 2011).
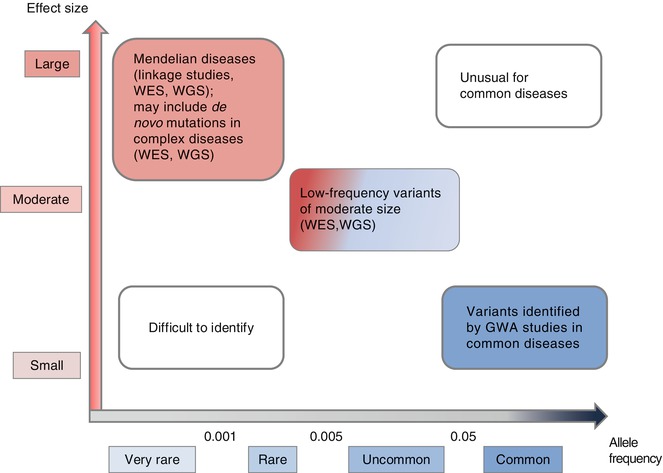
Fig. 5.1
Feasibility of gene identification studies by allele frequency and effect size in Mendelian and complex human diseases. Rare variants that have a large effect on the phenotype in Mendelian diseases can be identified in linkage studies where one investigates the segregation of a genetic marker with the disease phenotype in large multiple families affected with the disease. Very rare Mendelian diseases that are present in few small families are not amenable to this kind of linkage analysis and can only be identified if one sequences the whole exome (WES) or the whole genome (WGS) of the few affected individuals in order to identify the mutation specific to the phenotype. Recent genomics studies have implicated the presence of de novo (new) mutations in complex diseases that can only be identified by WES and/or WGS to identify the new mutation present in the affected individual and absent in parents. On the other end of the spectrum, common variants that have a small effect on the phenotype in complex diseases are identified by genome-wide association studies where one determines the association of a marker allele with the phenotype in a large cohort of cases and controls. Abbreviations: WES whole exome sequencing, WGS whole genome sequencing, GWA genome wide association
Gene identification studies of Chiari type I malformation and syringomyelia have been hindered by their complex aetiologies and inheritance patterns. Two approaches have been adopted or suggested in an attempt to identify the responsible genes and the underlying molecular pathogenic mechanisms. These are candidate gene studies and genome-wide linkage studies.
5.5.1 Candidate Genes Studies
A number of biologically plausible candidate genes, derived from mouse models, have been proposed for Chiari type I malformation, including the Hox genes , Pax genes , FGFR2 and Noggin . The Hox gene family controls the development of the occipital bone and ectopic expression14 of Hox–2.3 results in dysplasia or deficiency of occipital, basisphenoid and atlas bones in transgenic15 mice (McLain et al. 1992). The Pax group of genes codes for transcription factors with a conserved DNA-binding domain that have important roles in mesodermal segmentation and vertebral development. In particular, Pax1 plays an important role in somitic segmentation and proper sclerotomal differentiation in the cervico-occipital transitional zone (Chi and Epstein 2002). FGFR2 is transmembrane protein conserved across evolution and known to be critical for the normal development of multiple organ systems, including the craniofacial skeleton. The most common cause of Crouzon syndrome, a well-known craniosynostosis, is a mutation in FGFR2, and Chiari type I malformation is a common feature of Crouzon syndrome (Park et al. 1995). Noggin is required for growth and differentiation of the somites of the paraxial mesoderm (see Chap. 4). Noggin knockout mice show various defects, affecting neural and axial skeletal defects (McMahon et al. 1998). Noggin was analysed in 33 cases of Chiari type I malformation but no variant was identified, which suggests that this gene is not a common genetic factor involved in Chiari type I malformation (Speer et al. 2003).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


