Fig. 15.1
Acute hyperammoniemia resulting from liver failure results in astrocyte swelling. A representative electron micrograph showing swelling of a perivascular astrocyte (Ast) from a patient with acute liver failure who died of brain herniation. (Figure adapted from Felipo and Butterworth (2002) and reproduced with permission from Elsevier)
In contrast to ALF, end-stage chronic liver failure (cirrhosis) results in characteristic pathological changes to the astrocyte; these changes are known as Alzheimer type 2 astrocytosis where the cell nuclei take on a characteristic shape and pattern consisting of marked pallor and swelling with a prominent nucleolus, margination of the chromatin pattern and glycogen deposition (Fig. 15.2). The nuclei may take on an array of shapes and forms depending upon the brain structure examined. These forms range from the classical spherical shape in the cerebral cortex to irregular lobulated forms in some basal ganglia structures; the appearance of multiplets has been reported to suggest an element of hyperplasia (Norenberg 1987) .
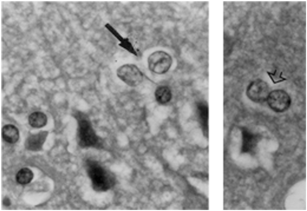
Fig. 15.2
Chronic hyperammoniemia resulting from liver cirrhosis results in Alzheimer type II astrocytes. Alzheimer type II cells (arrow, left-hand panel) show enlarged nuclei and margination of chromatin in this hematoxylin and eosin stained section of frontal cortex from a 51-year-old cirrhotic patient who died in hepatic coma. Doublets suggestive of a proliferative response are frequently seen (right-hand panel, arrow head). (Figure adapted from Felipo and Butterworth (2002) and reproduced with permission from Elsevier)
The occurrence of multiple episodes of grade 4 (or stage IV) HE (coma) and/or a single prolonged period of coma in a patient with end-stage chronic liver failure may result in a condition known as “acquired non-Wilsonian hepatocerebral degeneration”, the neuropathological features of which include Alzheimer type 2 astrocytosis . In addition, varying degrees of neuronal cell loss occur in cerebral cortex, cerebellum and basal ganglia structures (Butterworth 2007) . A related condition known as “Parkinsonism in cirrhosis” appears to result from manganese deposition in substantia nigra of these patients (Butterworth 2013b) .
Evidence of discrete alterations of the blood-brain barrier in HE have been proposed based upon studies of barrier permeability in an animal model of ALF (Nguyen 2010) but these findings were not confirmed by others (Bemeur et al. 2010a) . Swelling of cerebrovascular endothelial cells has been demonstrated using electron microscopy in material from ALF patients (Kato et al. 1992) again suggesting that, if barrier changes do occur in HE, they are discrete in nature.
Microglial activation and its role in the pathogenesis of HE was first described in the brains of animals with ALF resulting from hepatic devascularisation (Jiang et al. 2006) . Subsequent studies in this model revealed that activation of microglia was accompanied by grade 4 HE (coma) and brain edema as well as by increased expression of genes coding for pro-inflammatory cytokines in brain (Jiang et al. 2009a, b) (Fig. 15.3). Microglial activation has subsequently been confirmed using various techniques and cellular markers in an animal model of ALF resulting from toxic liver injury (McMillin et al. 2012b; Rangroo Thrane et al. 2012) . Activation of microglia has also been reported in material from a patient with ALF resulting from viral hepatitis (Butterworth 2011) , in a material from cirrhotic patients who died in hepatic coma (Zemtsova et al. 2011) and increased signals in positron emission tomography (PET) studies in HE patients was attributed to microglial activation (Cagnin et al. 2006) . Microglial activation has also been described in an animal model of biliary cirrhosis (D’Mello et al. 2009) .
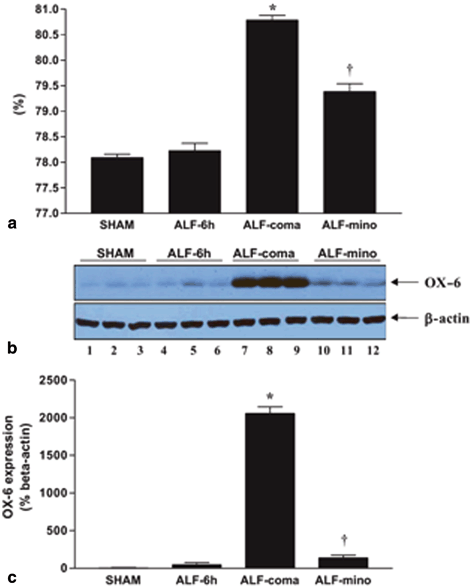
Fig. 15.3
Western blot analysis of OX-6 expression in acute liver failure (ALF) rats reveals that inhibition of microglial activation by minocycline treatment is correlated with attenuation of brain edema. a Percentage of brain water content of cerebral cortex from sham-operated controls (sham), ALF rats 6 h post- hepatic artery ligation/HAL (ALF-6 h), ALF rats at coma stage of encephalopathy (ALF-coma) and in ALF rats treated with minocycline (ALF-mino); b OX-6 protein expression in cerebral cortex from sham-operated controls (lanes 1–3), ALF rats 6 h post-HAL (lanes 4–6), ALF rats at coma stage of encephalopathy (lanes 7–9) and in ALF rats treated with minocycline (lanes 10–12); c Histogram representation of OX-6 expression in the various treatment groups. Data represent mean ± SEM of n = 10 animals per treatment group. Significant differences indicated by *p < 0.01 versus shan-operated controls and ALF-6 h; †p < 0.001 versus ALF-coma (ANOVA with post hoc Tukey’s test). (Figure reproduced from Jiang et al 2009a, with permission from Wiley)
The subject of activation of microglia in HE and its relationship to pro-inflammatory mechanisms and the role of cytokines/chemokines in the pathogenesis of the CNS complications of liver failure are covered in-depth in a later section of this chapter.
15.2 Abnormalities of Neuroglial Function in Liver Failure
In addition to frank neuroglial pathology, both acute and chronic liver failure result in a wide range of alterations of neuroglial function including deficits in neuroglial cell volume regulation and neuroglial-neuronal metabolic trafficking of key intermediates as well as neuroinflammatory changes. Some of these changes appear to relate to exposure of neuroglia in situ to ammonia and manganese, two substances known to accumulate to toxic concentrations in brain in liver failure .
15.2.1 Neuroglial Cell Volume Regulation
In ALF, it is generally considered that failure of cell volume regulation in astroglia underpins the phenomenon of cytotoxic brain edema that so frequently leads to increased intracranial pressure, brain herniation and patient death. Various mechanisms have been proposed to explain this failure of neuroglial cell volume regulation in ALF that include as described in two subheadings below.
15.2.1.1 Osmotic Effects of Increased Intracellular Glutamine
Being devoid of a functional urea cycle, brain relies upon glutamine synthesis for the removal of excess ammonia and the enzyme responsible, glutamine synthetase (GS) has a predominantly, if not exclusively, astroglial localization. Brain and cerebrospinal fluid (CSF) ammonia and glutamine concentrations correlate well with severity of HE in both ALF (Clemmesen et al. 1999) and in end-stage cirrhosis (Laubenberger et al. 1997) . Based upon studies in cultured cortical astrocytes exposed to ammonia as well as in animal models of pure hyperammonemia (in the absence of liver failure) , it has been proposed that the accumulation of glutamine in brain in liver failure leads to an osmotic gradient that contributes to cell swelling. This hypothesis is supported by a report describing a protective effect of the GS inhibitor methionine sulfoximine on cell swelling and brain edema in hyperammonemic animals (Brusilow et al. 2010) . However, although inhibition of glutamine synthesis was beneficial under these conditions, the use of 13C-nucler magnetic responence spectroscopy failed to show a correlation between de novo synthesis of glutamine in brain with either the severity of HE or with the presence of brain edema in animals with ALF (Chatauret et al. 2003) suggesting the presence of additional/alternative factors.
15.2.1.2 Neuroglial Proteins Involved in Cell Volume Regulation
Alterations in expression of genes coding for key neuroglial proteins with suggested roles in cell volume regulation have been reported in experimental models of HE.
Aquaporin-4 (AQP-4) is highly expressed in astroglia , particularly in the endfeet that ensheath brain capillaries where it mediates transmembrane movement of water. An important role for the protein has been suggested to contribute to the pathogenesis of brain edema in a range of clinical situations. In relation to the cerebral complications of liver failure , increased AQP-4 expression was reported in cultured cortical astrocytes exposed to millimolar concentrations of ammonia that led to significant cell swelling (Rama Rao et al. 2003) . However, results in animal models of liver failure have so far given conflicting results; whereas HE and brain edema were accompanied by increased concentrations of AQP-4 in one model of ALF (Eefsen et al. 2010) , no such changes were reported in a subsequent study (Wright et al. 2010) .
In contrast to the equivocal nature of AQP-4 in relation to HE, progressive decreases in expression of the astroglial marker protein, glial fibrillary acidic protein (GFAP) have consistently been reported in both experimental animal models of liver failure and in patient material. In rats with ALF resulting from hepatic devascularisation, GFAP expression was decreased as a function of the increase in brain water (Belanger et al. 2002) . Selective decreases of GFAP were previously reported in several brain structures from cirrhotic patients with HE; these structures included cerebral cortex, thalamus and basal ganglia (Sobel et al. 1981) whereas no such changes were subsequently reported in cerebellum of similar patients (Kril et al. 1997) . There is evidence to suggest a role of ammonia in the pathogenesis of the loss of GFAP in liver failure, a proposal that is based upon the report of a loss of expression of GFAP in cultured astrocytes exposed to ammonia which resulted in significant cell swelling and destabilization of the GFAP molecule (Neary et al. 1994) . It was proposed that loss of expression of GFAP, given its importance as a cytoskeletal protein in astroglia, could lead to altered visco-elastic properties of the cell thus favouring cell swelling (Belanger et al. 2002) .
Significant increases in expression of the astroglial 45 kDa isoform of the facilitative glucose transporter GLUT-1 were reported in an animal model of ALF (Belanger et al. 2006) and, as for GFAP, a role for exposure to ammonia was suggested. In addition to its established role in the transport of glucose, it has been proposed that GLUT-1 acts as a water channel leading to the suggestion that upregulation of the protein in ALF may contribute to (or result from) the appearance of brain edema (Belanger et al. 2006) .
One report described significant losses in expression of Kir 4.1, an inwardly-rectifying potassium channel expressed in astroglial endfeet that may have a function, in part, in cell volume regulation, has been described in a rat model of ALF (Obara-Michlewska et al. 2011) .
15.2.2 Neuroglial Amino Acid Transporter Proteins
High affinity transport of amino acids is a major function of astroglia. Such transport mechanisms exist to maintain the supply of key intermediates required for cellular energy metabolism , synthetic processes and for the termination of action of neurotransmitters such as glutamate and γ-aminobutyric acid (GABA) that form the basis of the so-called “glial-neuronal metabolic interactions “. Astroglial glutamate transporters are essential components of the glutamate-glutamine cycle and are responsible for the removal of excess glutamate from the synaptic cleft. Expression of the sodium-dependent, high affinity glutamate transporters EAAT-1 and EAAT-2 (in human; in rodents GLAST and GLT-1, respectively) have been reported in animal models of both acute and chronic liver failure (Knecht et al. 1997; Suarez et al. 2000) resulting in increases in extracellular brain glutamate concentrations (Vaquero and Butterworth 2006) . It has been suggested that limitations in the availability of glutamate in astroglia could limit ammonia-removal capacity since glutamate is the obligate precursor for GS, and, if so, could limit the synthesis of glutamine.
Decreases in expression of the high affinity glycine transporter (GLYT-1) have been reported in cerebral cortical extracts from rats with ALF resulting from hepatic devascularisation (Zwingmann et al. 2002) . Given the predominantly astroglial localization of GLYT-1 in cerebral cortex, it was proposed that these changes might relate to increases in availability of extracellular glycine with the potential to activate the glycine neuromodulatory site on the N-methyl-D aspartate (NMDA) subclass of glutamate receptor. NMDA receptor activation has been proposed to explain the hyperexcitability and nitrosative stress that occur in ALF (Vaquero and Butterworth 2006) .
Following the cloning and characterization of high affinity glutamine transporters (small neutral amino acid transporters or SNATs) and given the consistent finding of increased brain glutamine in HE, a study was undertaken to assess SNATs in an animal model of ALF (Desjardins et al. 2012) . Coma/edema stages of encephalopathy were accompanied by a selective decrease in expression of SNAT-5. Given the astroglial localization of SNAT-5 (Cubelos et al. 2005) it was proposed that down-regulation of transporter expression in liver failure could result in effective “trapping” of glutamine in the cell, an action that is consistent with cell swelling due to glutamine accumulation in the astrocyte as had previously been widely proposed. Moreover, since astroglial glutamine functions as immediate precursor of releasable (transmitter) glutamate, a limit upon its availability following decreased release from the astroglial cell has the potential to result in impairment of glutamatergic transmission an action that could result in excessive inhibition that is also characteristic of HE.
15.2.3 Neuroglial Translocator Protein
Translocator protein (TSPO), previously known as the “peripheral-type benzodiazepine receptor” is a mitochondrial protein responsible for the transport of cholesterol into the mitochondrion. The protein is expressed predominantly by neuroglia with both astroglia and microglia exhibiting high levels of expression in mammalian systems. Increased expression of TSPO has been reported in a wide range of hyperammonemic disorders including urea cycle enzymopathies, ALF, animals with end-to-side portacaval anastomoses and in patients with end-stage chronic liver failure (see (Ahboucha and Butterworth 2008) for review). In one study in humans using PET and the TSPO ligand 11C-PK11195, increased signals were observed in anterior cingulate cortex where the magnitude of the increased signals correlated with the degree of cognitive impairment (Cagnin et al. 2006) . The origin of these increased signals was considered to relate to microglial activation . Interest in TSPO in HE relates to the well-established relationship of the protein to the GABA system. Activation of neuroglial TSPO results in increased transport of cholesterol into the mitochondrion followed by increased synthesis of so-called “neurosteroids”, one of which, allopregnanolone is a very high affinity agonist for the neuronal GABA-A receptor. Increases in concentration of allopregnanolone were reported in autopsied brain tissue from patients who died in hepatic coma (Ahboucha and Butterworth 2005) . Based upon these findings, it was proposed that the increase in “GABAergic tone” resulting from activation of TSPO sites and the subsequent increase in synthesis of allopregnanolone could also contribute to the excessive neuroinhibition in HE.
15.3 Evidence for Microglial Activation in the Pathogenesis of HE
As stated above, microglial activation has been demonstrated to be a key feature in the pathogenesis of HE due to both ALF and chronic liver cirrhosis. Clinically, indirect evidence for microglial activation has been demonstrated by an upregulation of the microglial marker Ionized calcium binding adaptor molecule 1 (Iba-1) in post mortem cortical brain tissue from patients with liver cirrhosis and HE, when compared to cirrhotic patients without HE (Zemtsova et al. 2011) . In addition, data from a comprehensive gene expression profile analysis demonstrated an upregulation of markers for both the pro-inflammatory M1 and anti-inflammatory M2 microglial phenotypes, suggesting that both subpopulations of microglia may be present in patients with HE due to cirrhosis (Gorg et al. 2013) . Furthermore, increased [11C]-PK11195 binding to the TSPO in patients with proven cirrhosis and minimal HE was suggested to be a reflection of microglial activation in these patients (Cagnin et al. 2006) . Taken together, these clinical data indirectly support a role of microglia activation in HE.
In contrast, evidence for a direct role for microglia activation in the neurological consequences of both ALF and liver cirrhosis is more striking in many animal models of these diseases. Furthermore, in many of the models used, treatment modalities shown to inhibit microglia activation also alleviated or prevented the cognitive impairment and neurological decline observed during HE. Specific details are described below.
15.3.1 Toxic Liver Injury
A range of hepatotoxic agents have been used to uncover basic mechanisms responsible for the CNS complications of liver failure . This topic was reviewed by a panel of experts nominated by The International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) who, after careful deliberation, recommended two toxic models based upon the extent of the characterization. The two models of ALF were the azoxymethane (AOM) mouse model and the thioacetamide (TAA) rat model (Butterworth et al. 2009) . The AOM mouse model of ALF exhibits many of the pathophysiological characteristics of human HE due to acute liver failure . These features include (1) a clear pattern of neurological behaviors starting with the prodromal phase due to liver failure, where neurological symptoms are not yet evident followed by a number of distinct phases of neurological decline that rapidly progress to stupor andcoma; (2) the presence of cerebral edema; and (3) high levels of ammonia in the blood and brain. Very elegant and detailed analyses of the morphological changes in microglia and real-time analysis of microglial dysmotility after AOM have been demonstrated (Rangroo Thrane et al. 2012) . Both microglia activation (as demonstrated by an ameboidal phenotype) and motility (as demonstrated by analysis of the turnover rate) were shown to be altered in the cerebral cortex at late stages of HE when severe neurological symptoms were evident, coinciding with the appearance of brain edema (Fig. 15.4). Increased OX-42/CD11b immunoreactivity was also demonstrated in the cerebral cortex of AOM-injected mice (Chastre et al. 2012) , which was attenuated by treatment with the tumor necrosis factor (TNF)-α neutralizing molecule etanercept. Furthermore, there was a concomitant attenuation of AOM-induced liver injury and decreased expression of neuroinflammatory molecules in the brain after etanercept treatment (Chastre et al. 2012) .
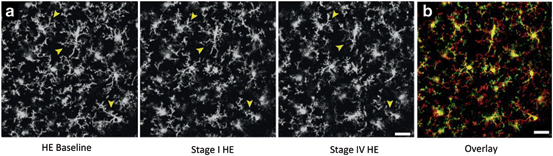
Fig. 15.4
The terminal coma stage of HE is associated with microglial activation. a Representative 2 photon laser scanning microscopy images from transgenic mice expressing green fluorescence protein under the control of the CX3CR1 promoter (expressed only in microglia) following AOM administration at 0 h (baseline), stage I and stage IV of HE (collapsed Z-stacks). b Overlay of representative images from HE at 0 h (red) and stage IV (green). Yellow represents the overlapping staining. During the latter stage, microglia appear more ameboid and activated. Scale bar represents 15 μM. (Figure adapted from Rangroo Thrane et al. (2012) and reproduced with permission from Elsevier)
15.3.2 Ischemic Liver Failure
Ischemic liver failure, although uncommon, is encountered clinically. Experimental ALF can be induced by the performing an end-to-side portacaval anastomosis followed by hepatic artery ligation. Rats undergoing this surgery exhibit key clinical features of HE, including cerebral edema and hyperammonemia, which ultimately result in grade 4 HE (hepatic coma). An increase in the number of OX-42/CD11b positive microglia has been demonstrated in the frontal cortex, thalamus and hippocampus starting 6 h after surgery (early stage HE) and worsening at the time of coma/edema (Jiang et al. 2009a, b), which could be alleviated by either mild hypothermia (Jiang et al. 2009b) or minocycline (Jiang et al. 2009a), with a concomitant attenuation of the progression of HE and brain edema .
15.3.3 Portal-Systemic (Bypass) Encephalopathy
In a related, more subtle model of HE induced by end-to-side portacaval shunt surgery alone, without subsequent hepatic artery ligation, rats develop mild cognitive impairment over the following 3–4 weeks. Associated with this mild form of HE, currently referred to clinically as “minimal HE”, is a change in the morphology of major histocompatibility complex class II (MHCII)-positive microglia to a more ameboid, activated phenotype (Agusti et al. 2011) . Curiously, these changes were restricted to cerebellum. Chronic infusion of a p38 mitogen-activated protein kinase inhibitor reversed the morphological changes observed in microglia and prevented the cognitive impairment (Agusti et al. 2011) .
15.3.4 Biliary Cirrhosis
Obstruction of the common bile duct induces a reproducible model of biliary cirrhosis in rats. Bile duct-ligated (BDL) animals have liver failure , developing jaundice, portal hypertension, portal-systemic shunting, bacterial translocation and immune system dysfunction. BDL rats are hyperammonemic but show only low-grade encephalopathy (decreased locomotor activities) (Butterworth et al. 2009) . Using this model, microglia are activated predominantly in the cerebellum, with only traces of activation in the striatum and thalamus (Rodrigo et al. 2010) (Fig. 15.5a). Interestingly, no microglia activation was evident in the frontal cortex. Treatment with ibuprofen reduced the microglia activation and reversed the concomitant cognitive impairments observed (Rodrigo et al. 2010) . Similarly, microglia activation has been shown after BDL in mice, as demonstrated by morphological changes in Iba-1 positive microglia (D’Mello et al. 2009) . However, in contrast to the rat model, the activation of microglia was localized to the cerebral cortex rather than the cerebellum (Fig. 15.5b). The cause of these region-and species-selective changes remains unknown. As discussed in a separate section of this chapter, the activation of microglia in BDL mice is thought to subsequently recruit monocytes to the brain that contribute to the cognitive impairment observed (Kerfoot et al. 2006) .
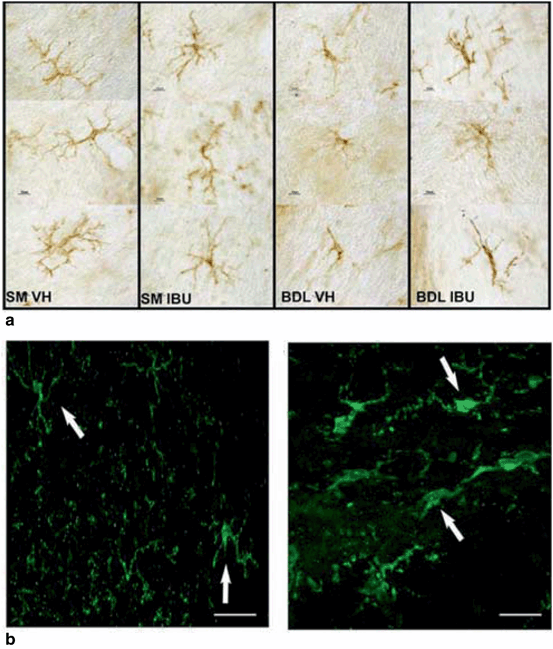
Fig. 15.5
Experimental models of biliary cirrhosis and mild HE are associated with microglial activation. a Rats underwent bile duct ligation (BDL) or sham (SM) surgery and were subsequently treated with either treated with vehicle (VH) or Ibuprofen (IBU). Microglial activation was assessed using MHCII expression. Microglia in the cerebellum of BDL rats showed an activated, ameboid phenotype. (Figure adapted from Rodrigo et al. (2010), and reproduced with permission from Elsevier.) b Immunohistochemistry staining for Iba-1-positive microglia in the cortex of BDL mice. Microglia in mice undergoing sham surgery (left panel) have resting ramified morphology and longer processes, while after BDL, microglia are more rounded with retracted and thicker processes indicative of microglia activation. (Figure adapted from D’Mello et al. (2009) and reproduced with permission from the Society for Neuroscience)
15.3.5 Ammonia Neurotoxicity
It has long been accepted that ammonia plays an important role in the neurological symptoms of HE. Hyperammonemia has been demonstrated in the blood and cerebral cortex at all stages of HE in both ALF and in cirrhosis. Furthermore, injection of a bolus dose of ammonium acetate into rats in the absence of liver failure results in a transient comatose state, supporting a role for ammonia in the neurological decline of HE. In contrast, rats fed an ammonium-containing diet for up to 28 days display cognitive deficits more indicative of minimal HE (Felipo et al. 1988) . However, the role of ammonia toxicity in the activation of microglia and subsequent neuroinflammatory response is not as clearly defined. Treatment of primary microglial cultures with ammonia results in microglia swelling, migration , alterations in filipodia length, and Iba-1 expression (Zemtsova et al. 2011; Lachmann et al. 2013) . Similarly, increased Iba-1 immunoreactivity and expression was demonstrated in the cerebral cortex of the in vivo rat model of ammonia intoxication (Zemtsova et al. 2011). In contrast, injection of an acute bolus dose of ammonium acetate into mice had no effect on the morphology or turn-over rate of microglia (Rangroo Thrane et al. 2012) .
Microglial activation was evident in the cerebellum of chronic ammonium-fed rats, as demonstrated by in-depth morphometric analyses of MHCII positive cells (Rodrigo et al. 2010) . In support of a role for microglia activation in the cognitive deficits observed in this chronic hyperammonia model, treatment with ibuprofen reduced the microglia activation and reversed the concomitant cognitive impairments observed (Rodrigo et al. 2010) .
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Ionic Signaling in Physiology and Pathophysiology of Astroglia
Ionic Signaling in Physiology and Pathophysiology of Astroglia
 Amyotrophic Lateral Sclerosis: A Glial Perspective
Amyotrophic Lateral Sclerosis: A Glial Perspective
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Novel Therapeutic Approaches to Malignant Gliomas
Novel Therapeutic Approaches to Malignant Gliomas
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


