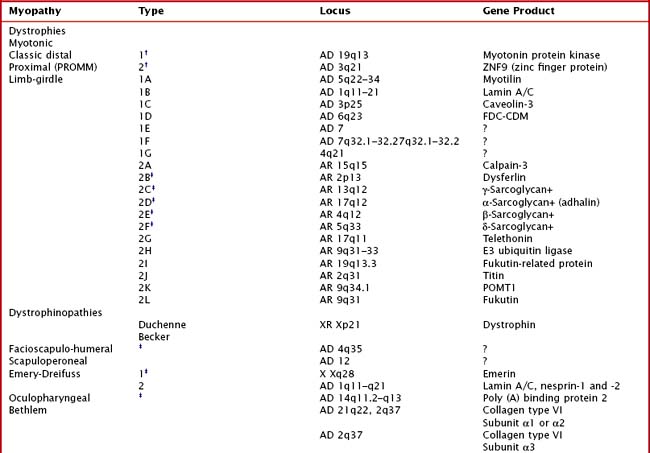
75 Hereditary Myopathies
Hereditary muscle disorders are usually generalized muscle disorders that are progressive and are variously categorized as channelopathies, metabolic and mitochondrial myopathies, muscular dystrophies, and congenital myopathies (Table 75-1).


Channelopathies
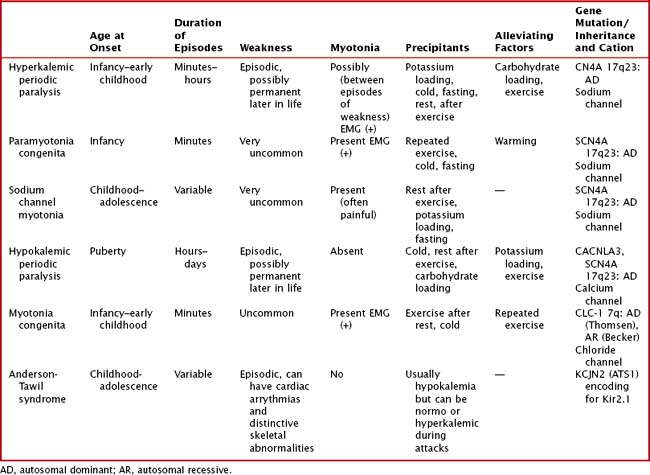
Periodic Paralysis and Congenital Myotonic Disorders
Clinical Vignette
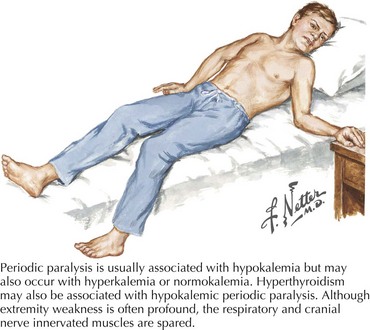
Variable mutations within genes encoding muscle membrane ion channels are responsible for the different forms of periodic paralyses as well as other myotonic disorders (Table 75-2). Most of these patients have an autosomal dominant inheritance. During the episodic paralyses, the skeletal muscle membrane excitability transiently disappears. The degree of weakness may vary from one family member to another; boys and men are more often significantly affected.
Clinical Presentation
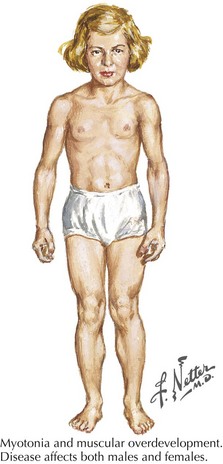
Myotonia congenita, a chloride channelopathy, is especially aggravated by immobility and ameliorated by exercise and warming; here the myotonia per se is easily elicited on examination. Fixed weakness is not usually present in dominant myotonia congenita. A rather unique characteristic of Thomsen disease variant is the pseudo-hypertrophy of the skeletal muscles providing the patient with a rather pseudo-herculean habitus (Fig. 75-2). It may be so profound that athletic coaches enthusiastically encourage these individuals to participate in sports activities. Unfortunately, some of these individuals may develop a mild progressive weakness. Transient episodes of true weakness precipitated by sudden movements after rest that are relieved by exercise are characteristic of myotonia congenita. Interesting examples include a baseball player who cannot run after hitting the ball or a subway rider who wishes to get off when the train stops but is frozen in place or falls when he arises to leave the train.
Differential Diagnosis
Myasthenia gravis (MG) classically has a remitting relapsing course predominantly confined to ocular as well as bulbar muscles and to a lesser extent proximal extremity musculature. MG is typified by fluctuating, rarely acute weakness with a very definitive diurnal variation (see Chapter 73).
Glycogen and Lipid Storage Disorders
Clinical Vignette
Comment: This is a classic example of muscle phosphorylase deficiency (Fig. 75-3) with onset in adolescence when individuals for the first time have the muscle power to allow them to stress their metabolic system to the point of actual muscle necrosis and subsequent myoglobinuria, the feature that most commonly brings them to medical attention.
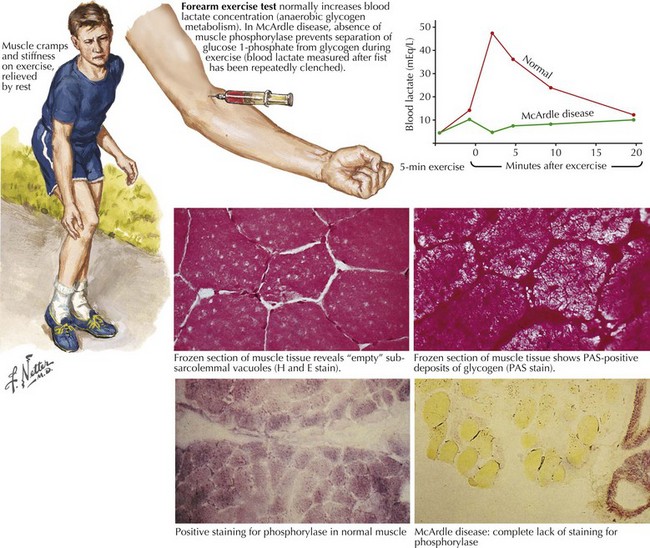
Glycogen storage disorders (GSDs) are very uncommon clinical entities. The classic picture is one of exercise-induced painful muscle cramps, associated with myoglobinuria. The concomitant laboratory documentation of profound elevated serum CK levels and myoglobinuria strongly implicates either a carbohydrate or lipid enzymatic deficiency of inborn metabolism (Fig. 75-4; Table 75-3). Very rarely prolonged use of one extremity, in isolation, will uncover the presence of a previously unsuspected glycogen storage disease (GSD). Muscle phosphorylase deficiency, an inborn error of glycogen metabolism, is the most common of these GSD myopathies.
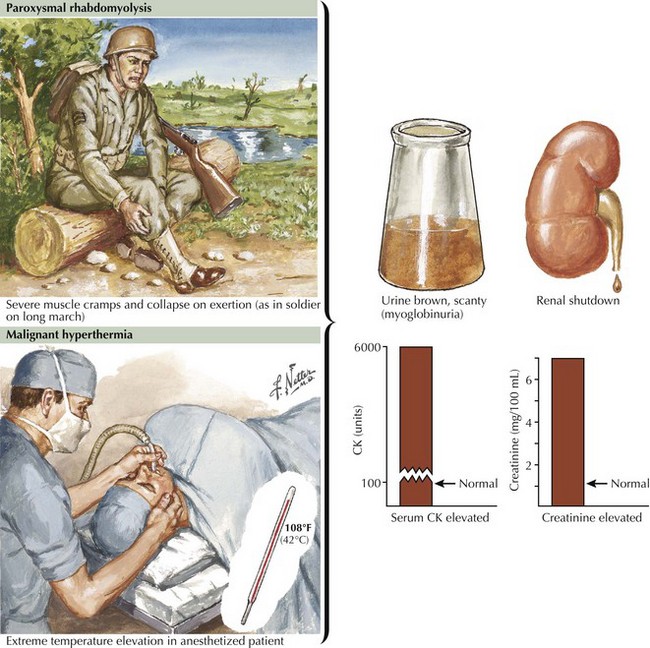
Table 75-3 Myopathies Presenting with Exercise Intolerance
| Glycogenoses | Respiratory Chain Defects | Lipid Metabolism Disorders |
|---|---|---|
| Myophosphorylase deficiency (McArdle disease)—Type V | Complex 1 deficiency | Carnitine deficiency |
| Phosphofructokinase deficiency (Tauri disease)—Type VII | Coenzyme Q10 deficiency | Carnitine palmitoyltransferase deficiency |
| Phosphorylase B kinase deficiency—Type VIII | Complex III deficiency | Very long chain, long chain, medium chain, or short chain acyl CoA dehydrogenase deficiency |
| Phosphoglycerate kinase deficiency—Type IX | Complex IV deficiency | 3-Hydroxy Acyl-CoA dehydrogenase deficiency protein deficiency |
| Phosphoglycerate mutase deficiency—Type X | Complex V deficiency | Glutaric aciduria type II (electron-transferring flavoprotein and CoQ oxidoreductase deficiencies) |
| Lactate dehydrogenase deficiency—Type XI | Combination of I to V | Neutral lipid storage disease with myopathy; neutral lipid storage disease with ichthyosis |
| Beta enolase deficiency—Type XII |
Pathophysiology
Skeletal muscle function is extremely energy dependent. Normal muscle metabolism requires the presence of both circulating glucose and free fatty acids (Figs. 75-5 and 75-6). At rest, muscles use fatty acids for basal metabolic demands. When one first begins to vigorously exercise, usually within the first 10 minutes, the glycolysis of glycogen, already stored within muscle tissues, is the primary energy source as its breakdown produces glucose but for a relatively short time period. However, when the vigorous exercise is prolonged past these first few minutes, the body shifts to anaerobic glycolysis. This is manifested clinically by the second wind phenomenon. Here lipid stores, in the form of free fatty acids, are mobilized as the primary source of energy. Effective glycolysis is blocked in the various muscle glycogenoses. This essentially deprives muscle of the initial need for glucose, and consequently an accumulation of underutilized glycogen occurs within muscle. In essence, that is, these muscles are inappropriately stressed by what for most healthy persons is no more than strenuous exercise.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


