Hydrocephalus
The surgical management of hydrocephalus has undergone fantastic changes over the last generation of neurosurgeons: dramatic improvements in imaging with computed tomography (CT) and magnetic resonance (MR) imaging, remarkable innovative advances in cerebrospinal fluid (CSF) valve technology, bewildering complex computer models, and dramatic advances in endoscopic equipment and techniques. In terms of overall patient outcomes, one could conclude, however, that things are a little better, but “not much.” This frustrating yet fascinating dichotomy between technological advances and clinical outcomes makes hydrocephalus, first described by the ancients, one of the most understated and complex disorders. For pediatric neurosurgeons, it is the disorder that they most commonly treat. The challenge to the current generation of pediatric neurosurgeons is to solve this vexing problem, through a better understanding of basic science, better computer models, further technological advances, and most importantly a broad-based, concerted mult-disciplinary attack on this disorder. This chapter focuses on an overview of pediatric hydrocephalus. Further chapters will address its management with CSF shunts and endoscopy.
9.1 Definition
Hydrocephalus is possibly most simply defined as an increase in the fluid-containing spaces of the brain at increased pressure, resulting from an imbalance between CSF production and absorption or flow. This definition excludes other abnormalities of CSF dynamics, such as benign intracranial hypertension, in which the ventricles are not enlarged, and hydrocephalus ex vacuo, in which cerebral atrophy and focal destructive lesions lead to an abnormal increase of CSF passively. However, not all cases fall clearly within these definitions; patients with parenchymal destruction may develop progressive hydrocephalus, and patients with slit-ventricle syndrome may have a presentation akin to that of benign intracranial hypertension.
9.2 Classification
There is no universally accepted classification system for hydrocephalus. A number have been suggested,1–5 which include the following:
Communicating versus noncommunicating
Obstructive versus absorptive
Acquired versus congenital
Genetic or central nervous system (CNS) malformation–associated versus isolated
Intraventricular–obstructive versus extraventricular
Simple versus complicated
The terms communicating and noncommunicating date back to the early 1900s and are based on the experimental studies of Dandy and Blackfan,6 who investigated the pathophysiology of hydrocephalus in which the aqueduct is obstructed. Communicating implies that the CSF has free flow into the subarachnoid space, but in the absence of overproduction, it is generally accepted that CSF absorption is impaired (or obstructed) downstream at some point. The terms compensated hydrocephalus and “arrested” hydrocephalus are also older and generally refer to whether an increase in ventricular size is associated with evidence of raised intracranial pressure (ICP). In some cases, the gradual increase in ventricular size stabilizes by reaching a new equilibrium, and the patient has no symptoms or signs of raised ICP. In others, a patient is discovered incidentally to have large ventricles unassociated with any identifiable problems. However, patients with apparently arrested hydrocephalus may still develop symptoms and signs at a later date, so the process is not entirely static.7 Congenital hydrocephalus is present at birth and often is associated with developmental defects; acquired hydrocephalus occurs after development of the brain and ventricles.4,8 Hydrocephalus has also been classified based on the stage of development at the time that the ventricles became dilated.9 The various subtypes of fetal hydrocephalus are classified according to the mechanism of obstruction to the flow of CSF; they include primary or simple hydrocephalus, with a single point of obstruction to flow; dysgenetic hydrocephalus, with complex abnormalities of the CNS, such as Arnold-Chiari malformation; and secondary hydrocephalus resulting from tumor or bleeding. This classification is cross-referenced to the stage of fetal development (i.e., neuronal maturation, cell migration). This classification may prove useful in deciding when treatment may be futile if the legal period for terminating a pregnancy has elapsed and in identifying potential candidates for early delivery or fetal surgery.5
Extraventricular obstructive hydrocephalus is now recognized to represent, almost universally, benign pericerebral collections of infancy that are usually familial, resolve with time, and almost never require treatment.10 Exceptions include genetic conditions, such as certain mucopolysaccharidoses, achondroplasia, Sotos syndrome, and glutaric aciduria type 1, which are often associated with developmental delay. In these cases, identification is important, as therapeutic options exist for many forms of mucopolysaccharidosis and for glutaric aciduria type 1.11
9.3 Epidemiology
The exact incidence of hydrocephalus is hard to ascertain as it is generally secondary to some other insult, including tumor, infection, trauma, or prematurity. Cited rates in newborns range from 0.3 to 4 per 1,000 live births. When it occurs as a single congenital disorder, the incidence of hydrocephalus has been reported as 0.9 to 1.5 per 1,000 births.12–16 One estimate suggested that approximately 125,000 persons are living with CSF shunts and that 33,000 shunts are placed annually in the United States.17
The incidence of pediatric hydrocephalus has declined in many developed countries.10 Antenatal screening, genetic testing, and pregnancy termination have reduced the incidence of the congenital malformations of the brain that cause hydrocephalus. The incidence of open neural tube defects has also decreased precipitously as a result of maternal folate supplementation, antenatal screening, and termination of pregnancy based on superior antenatal imaging with ultrasound and MR imaging. The incidence of CSF shunting in open neural tube defects, formerly reported to be as high as 90%, has also declined, possibly as a result of a generally more conservative approach, and the selection of anatomically lower lesions for delivery with a lower requirement for shunting.18,19 The incidence of intraventricular hemorrhage (IVH) has decreased as a result of better perinatal management of prematurity.20
9.4 Cerebrospinal Fluid Production, Circulation, and Absorption
CSF is produced by two mechanisms. Most of the CSF (50 to 80%) is thought to be secreted by the choroid plexus within the cerebral ventricles. Extrachoroidal CSF production in subarachnoid sites and by way of a transependymal route has also been documented. About 20% or more of the CSF is derived from brain extracellular fluid created as a by-product of cerebral metabolism.21–23 Normally, rates of production (0.35 mL/min or approximately 400 to 500 mL/d) and absorption of CSF are equal. Total CSF volume is 65 to 140 mL in children and 90 to 150 mL in adults
The process of CSF formation by the choroid plexus includes plasma ultrafiltration and secretion. Secretion, an energy-dependent process, is initiated by hydrostatic pressure in the choroidal capillaries and by active transport of sodium. The enzymes sodium–potassium adenosine triphosphatase and carbonic anhydrase partly regulate CSF secretion.23 CSF production has been reported to remain constant across the normal range of ICP,24 with CSF production decreasing when the ICP approaches mean arterial pressure. There have been reports, however, of downregulation of CSF production in patients with chronic hydrocephalus.25 In contrast to CSF production, CSF reabsorption is not an energy-dependent process.22 Information gained from MR imaging analysis of CSF movement demonstrates pulsatile to-and-fro motion of CSF within the lateral ventricles, produced from a brain-pumping motion that ejects the CSF and causes a net downward flow.26
Historically, it has been held that CSF is absorbed into the vascular system mainly through the arachnoid villi within the arachnoid granulations covering the brain and spinal cord leptomeninges.27,28 This process is thought to be passive and not energy-dependent. A layer of endothelium within the arachnoid villi separates the subarachnoid CSF space from the vascular system. Water and electrolytes pass freely across these arachnoid membranes. There is normally a 5- to 7-mm Hg difference in pressure between the dural venous sinuses and the subarachnoid space. This is presumed to be the hydrostatic force behind the absorption of CSF. Larger proteins and macromolecules cannot pass through intercellular junctions but are selectively transported across the cytoplasm of endothelial cells by an active process involving micropinocytosis.28 Increased absorption through the arachnoid villi protects the brain from transient increases in ICP.29 This concept has, however, been recently challenged. Newborn infants do not have visible arachnoid granulations yet maintain normal CSF circulation. Johnston et al30–32 have produced evidence in a number of animal species and human cadavers that the main route of CSF absorption is via the olfactory nerves, cribriform plate, and nasal lymphatics. At pressures above normal, the arachnoid granulations become active, perhaps explaining earlier experimental findings. Absorption of CSF across brain tissue into capillaries has also been proposed,33 and according to this theory, the distending force in the production of chronic hydrocephalus is an increased systolic pulse pressure in the brain tissue that is due to decreased intracranial compliance.
9.5 Etiology and Pathophysiology
Hydrocephalus can be a symptom of a large number of disorders: it is associated with tumors and infections and occurs as a complication of prematurity and trauma.34–36 It is also seen in apparent isolation. High-resolution MR imaging of postnatal life has provided clues to the etiologies of hydrocephalus, which in the past would have been labeled as idiopathic; some of these include IVH (▶ Fig. 9.1 A–C), aqueduct stenosis (▶ Fig. 9.2), and migrational abnormalities.10 The etiologies of hydrocephalus in one series of pediatric patients are shown in ▶ Table 9.1.
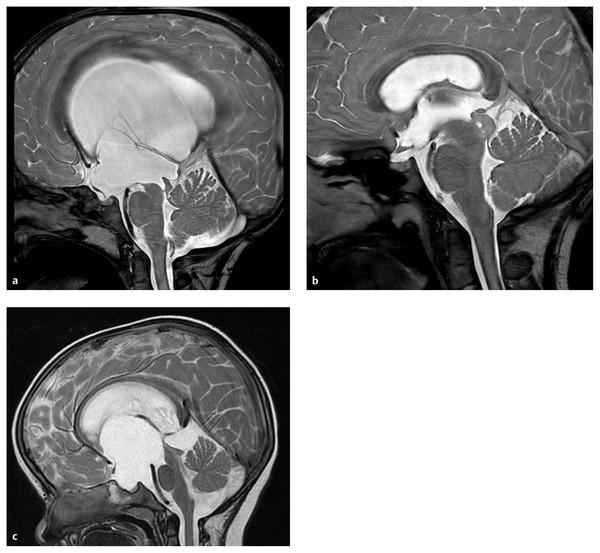
Fig. 9.1 Post-intraventricular hemorrhage (IVH) hydrocephalus. (a) Initial coronal ultrasound showing IVH with extension into parenchyma. (b) Axial magnetic resonance images at two different levels demonstrating slight enlargement of the ventricles. Note immature sulcal development. (c) Coronal ultrasound following progression of hydrocephalus that led to surgical treatment.
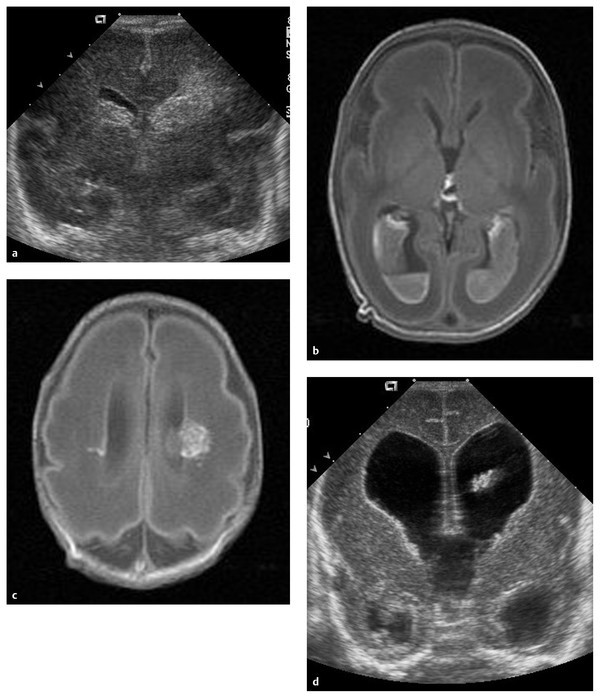
Fig. 9.2 Intrauterine magnetic resonance image of severe hydrocephalus. (a-d) Sequential coronal views.
| Causes | Percentage of patients |
| Intraventricular hemorrhage | 24.1 |
| Myelomeningocele | 21.2 |
| Tumor | 9.0 |
| Aqueductal stenosis | 7.0 |
| Infection | 5.2 |
| Head injury | 1.5 |
| Other | 11.3 |
| Unknown | 11.0 |
| Two or more causes | 8.7 |
| Source: Modified with permission from Drake et al, 1998. | |
Hydrocephalus is due to abnormal CSF reabsorption, flow, or rarely overproduction.
The main situation in which CSF production is increased enough to cause hydrocephalus is the presence of a choroid plexus papilloma. These tumors contain functional choroid epithelium and can produce very large amounts of CSF. However, even in such cases, reabsorption is probably defective because otherwise-normal individuals can usually tolerate the elevated CSF production rate of these tumors. CSF accumulation, in turn, leads to raised ICP.
The etiology of hydrocephalus depends upon the age of the child. During the neonatal period to late infancy (0 to 2 years), hydrocephalus is usually caused by perinatal hemorrhage, meningitis, or developmental abnormalities, the most common being aqueductal stenosis. The hydrocephalus seen in babies with spina bifida usually results from an associated Chiari malformation.
In early to late childhood (2 to 10 years), the most common causes of hydrocephalus are posterior fossa tumors and aqueduct stenosis.
9.5.1 Congenital Causes in Infants and Children
Approximately 55% of all cases of hydrocephalus are congenital. Primary aqueductal stenosis accounts for approximately 5% of cases of congenital hydrocephalus, whereas aqueductal stenosis secondary to neoplasm, infection, or hemorrhage accounts for another 5%.34 Primary aqueduct stenosis usually presents in infancy. Its morphology may be that of “forking” of the aqueduct, an aqueductal septum, “true” narrowing of the aqueduct, or X-linked aqueduct stenosis. Bickers-Adams syndrome is an X-linked form of hydrocephalus accounting for 7% of cases in males. It is characterized by stenosis of the aqueduct of Sylvius, severe mental retardation, and in 50% of cases by an adduction–flexion deformity of the thumb. Secondary aqueduct stenosis is due to gliosis secondary to intrauterine infection or germinal matrix hemorrhage.37
Anatomical malformations frequently observed with idiopathic congenital hydrocephalus include Chiari malformations, Dandy-Walker malformation, and others.36,38 Dandy-Walker malformation is associated with atresia of the foramina of Luschka and Magendie and affects 2 to 4% of newborns with hydrocephalus. About 50% of all patients with Dandy-Walker malformation develop hydrocephalus. The dilated fourth ventricle does not communicate effectively with the subarachnoid space. In Chiari malformations, hydrocephalus may occur with fourth ventricle outlet obstruction in Chiari type 1 malformation and is commonly associated with myelomeningocele in Chiari type 2 malformation.
Hydrocephalus occurs in approximately 80 to 90% of patients with myelomeningocele; of these cases, 50% are obvious at birth.18,39
Neonatal hydrocephalus can also be part of a major cerebral malformation, such as an encephalocele or holoprosencephaly, or can be associated with inherited metabolic diseases, such as achondroplasia and Hurler disease. Other causes of congenital hydrocephalus include agenesis of the foramen of Monro, congenital tumors, arachnoid cysts and vascular malformations, and intrauterine toxoplasmosis.
9.5.2 Acquired Causes in Infants and Children
Infective causes of hydrocephalus include meningitis, especially bacterial, which can lead to hydrocephalus by either inflammatory aqueduct stenosis or leptomeningeal fibrosis. In some geographic areas, parasitic disease, such as intraventricular cysticercosis, can cause hydrocephalus by mechanical obstruction.
Post-hemorrhagic hydrocephalus occurs following IVH, which can be related to prematurity, head injury, or rupture of a vascular malformation. Communicating hydrocephalus after subarachnoid hemorrhage is more common in adults and is rarely seen in children. Over the past two decades, there has been remarkable improvement in the survival of extremely low-birth-weight infants; however, the most immature of these infants remain at increased risk for neonatal complications that potentially affect long-term neurologic developmental outcome, including IVH. The risk for severe IVH varies inversely with gestational age, with an overall incidence of 7 to 23%.40–42 Approximately one-third of extremely low-birth-weight infants with an IVH develop post-hemorrhagic hydrocephalus, and 15% of them will require shunt insertion.43–45
Hydrocephalus after IVH is usually ascribed to fibrosing arachnoiditis, meningeal fibrosis, and subependymal gliosis, which impair the flow and reabsorption of CSF. Recent experimental studies have suggested that acute parenchymal compression and ischemic damage and increased parenchymal and perivascular deposition of extracellular matrix proteins—probably due at least partly to upregulation of transforming growth factor-β (TGF-β)—are further important contributors to the development of the hydrocephalus. IVH is associated with damage to periventricular white matter, and the damage is exacerbated by the development of hydrocephalus; combinations of pressure, distortion, ischemia, inflammation, and free radical–mediated injury are probably responsible.46
Mass lesions account for 20% of all cases of hydrocephalus in children. These are usually tumors such as medulloblastoma, astrocytoma, and ependymoma, but cysts, abscesses, vascular malformations, or hematomas also can be the cause. Twenty percent of pediatric patients develop hydrocephalus requiring shunting following posterior fossa tumor removal. This may be delayed up to a year. Risk factors for the requirement of CSF diversion in this group include age younger than 2 years, papilledema, enlarged ventricles, presence of metastases, and tumor type.47 Increased venous sinus pressure can also lead to hydrocephalus. This can be related to achondroplasia, some forms of craniosynostosis, or venous sinus thrombosis.
Iatrogenic causes of hydrocephalus include hypervitaminosis A, which can lead to hydrocephalus by increasing secretion of CSF or by increasing permeability of the blood–brain barrier. Hypervitaminosis A is a more common cause of idiopathic intracranial hypertension.48,49
9.6 Clinical Characteristics
The clinical features of hydrocephalus depend on the age of the patient at presentation and the time of onset in relation to closure of the cranial sutures. With the current advances in antenatal monitoring, the majority of congenital cases of hydrocephalus are diagnosed early (▶ Fig. 9.3), allowing planned cesarian delivery in the moderate to severe cases in which cephalopelvic disproportion is expected.
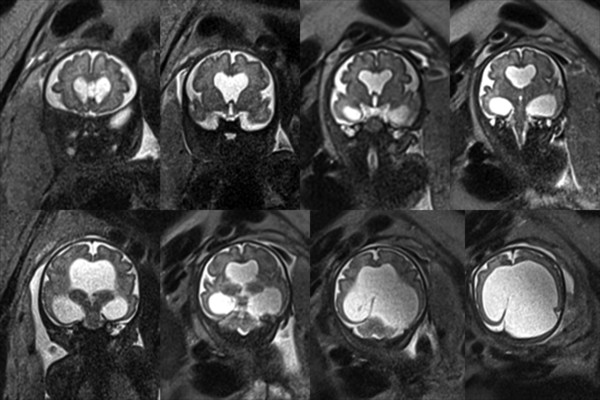
Fig. 9.3 Obstructive hydrocephalus from (a) aqueduct occlusion, (b) tectal glioma, and (c) suprasellar arachnoid cyst.
9.6.1 Symptoms and Signs in Infants
Hydrocephalus can present as acute raised ICP, but because of the relative distensibility of the infant skull, the presentation may be more subtle, with failure to thrive and achieve milestones. Infants with hydrocephalus may be drowsy and irritable. Poor feeding and vomiting are common. These infants may have apneic spells, bradycardia, and a bulging, tense anterior fontanel. Head circumference increases abnormally across centiles: the head circumference is at or above the 98th percentile for age. The scalp veins may be distended, the scalp skin thin and shiny, and the cranial sutures splayed. In the minority of cases, an abnormally large head is present at birth. In most, however, the hydrocephalus only gradually becomes obvious. In advanced cases, clinical examination reveals a significant craniofacial disproportion with expansion of the dome and low-set ears and eyes. In very severe cases, in which the cerebral cortex is thinned, transillumination of the cranial cavity may be possible. Epileptic seizures are rarely seen as a result of hydrocephalus alone.
Papilledema is rare in this age group, although funduscopy may reveal retinal venous engorgement. Oculomotor abnormalities may include abducens nerve palsy. Upgaze palsy from third ventricular pressure on the midbrain tectum, producing a “setting-sun” sign, can be observed, although this is usually absent in premature infants. In babies over the age of 6 months, limb tone may be increased, with spasticity preferentially affecting the lower limbs. However, some infants with definite hydrocephalus exhibit no such signs, as hydrocephalus may have developed slowly and the splaying of the sutures may have prevented the ICP from rising considerably. In fact, some infants with hydrocephalus after intraventricular hemorrhage may show hypotonia.
9.6.2 Symptoms and Signs in Older Children
In older children (older than 2 years), the head circumference is usually within normal limits if hydrocephalus develops after closure of the cranial sutures or may be increased in children with preexisting (infantile) but unrecognized progressive hydrocephalus.
Learning problems and reduced intellectual function are common, and neurologic development may be delayed. School-age children may have deteriorating school performance as a result of headaches, failing mental function, memory loss, or behavioral disturbances. More acutely, these children present with symptoms and signs of increased ICP, such as headache, nausea and vomiting, drowsiness, gait changes, papilledema, and upgaze and/or abducens palsy. Failure of upward gaze is due to pressure on the tectal plate through the suprapineal recess. The limitation of upward gaze is of supranuclear origin. When the pressure is severe, other elements of the dorsal midbrain syndrome (Parinaud syndrome) may be observed, such as light–near dissociation, convergence–retraction nystagmus, and eyelid retraction (Collier sign).
Evidence of abnormal hypothalamic function (e.g., short stature or gigantism, obesity, delayed puberty, primary amenorrhea or menstrual irregularity, and diabetes inspidus) may occur secondary to increased ICP or dilatation of the third ventricle. Difficulty in walking may occur secondary to truncal and limb ataxia or limb spasticity. This affects the lower limbs preferentially because the periventricular pyramidal tract is stretched by the enlarged ventricles.
Neck pain may indicate associated tonsillar herniation, and blurred vision may be present as a consequence of papilledema, which if left untreated leads to optic atrophy.
9.7 Genetics
Although commonly considered a single disorder, human hydrocephalus is a collection of heterogeneous, complex, and multifactorial disorders. A growing body of evidence suggests that genetic factors play a major role in the pathogenesis of hydrocephalus.50 Congenital hydrocephalus may occur alone (nonsyndromic) or with other anomalies (syndromic). It is estimated that about 40% of cases of hydrocephalus have a possible genetic etiology.51
In genetic terms, the isolated (nonsyndromic) form of hydrocephalus is a primary and major phenotype caused by a specific faulty gene. In syndromic forms of congenital hydrocephalus, it is difficult to define the defective gene because of the association with other anomalies. Autosomal-recessive, autosomal-dominant, X-linked recessive,52 and X-linked dominant53 forms of hydrocephalus are recognized.
At least 43 mutants/loci linked to hereditary hydrocephalus have been identified in animal models and humans. To date, nine genes associated with hydrocephalus have been identified in animal models, whereas only one such gene has been identified in humans: the hydrocephalus (X-linked) gene.51 Cases of X-linked hydrocephalus (HSAS1, OMIM) comprise approximately 5 to 15% of the congenital cases with a genetic cause.51,54
The gene responsible for X-linked human congenital hydrocephalus is at Xq28 and encodes L1CAM (L1 cell adhesion molecule).55 CRASH (corpus callosum agenesis, retardation, adducted thumbs, shuffling gait, and hydrocephalus) incorporates the L1 mutations that were formerly regarded as multiple separate entities.56 Congenital aqueductal stenosis can also be caused by autosomal-recessive mechanisms (OMIM 236635).57 In general, the recurrence risk for congenital hydrocephalus, excluding X-linked hydrocephalus, is low. Empiric risk rates range from less than 1% to 4%,58 indicating the rarity of autosomal-recessive congenital hydrocephalus.51,54,59 However, multiple human kindreds with congenital hydrocephalus have been reported.50,51,54,60
Hydrocephalus has been observed in many mammals.50 Review of the molecular etiologies shows a very diverse set of pathogenetic mechanisms. Perturbation of almost any molecule that plays a crucial role in early brain development and the sequential regulation of CSF dynamics may be involved. Hydrocephalus may also be caused by a malfunction of the ependymal cells,61 mesenchymal cells,62,63 growth factor signaling,64,65 disruption of extracellular matrix (ECM),66,67 and membrane fusion events (synaptosomal-associated protein, or SNAP).68
9.8 Imaging Studies
9.8.1 Cranial Ultrasound
Ultrasound is by far the quickest, cheapest, and most convenient method for demonstrating ventricular enlargement in infants with an open fontanel, particularly premature infants with post-hemorrhagic hydrocephalus, and for serial imaging in follow-up. It is also useful for the diagnosis of intrauterine hydrocephalus. Measurement of ventricular width from the midline to the lateral border of the lateral ventricle in the midcoronal view is the measurement with the least interobserver variability, and centiles for gestational age have been compiled.69
No sedation is required for acquiring of ultrasound images, and the procedure can be repeated frequently without any adverse effects. It may not visualize the posterior fossa well and may not always establish an etiologic diagnosis.
9.8.2 Computed Tomography
CT demonstrates the size and morphology of the ventricles and periventricular lucency, and it can reveal underlying pathologies, such as hemorrhage and posterior fossa tumors. Baseline imaging of asymptomatic patients (especially after shunt revision) may serve as a reliable study for comparison with subsequent imaging when patients become symptomatic. CT is widely available in many facilities and often does not require sedation of the child. However, there is mounting concern about the effects of exposure to radiation, so that alternate imaging70ultrasound or rapid-sequence MR imaging—is frequently preferred.71
9.8.3 Magnetic Resonance Imaging
MR imaging provides a better morphological definition and a better etiologic diagnosis, such as the presence of low-grade gliomas or colloid cysts, which may not be demonstrated on CT. It is better for evaluating Chiari malformations and cerebellar or periaqueductal tumors. Cine MR imaging is an MR imaging technique to measure CSF stroke volume in the cerebral aqueduct. This is used for demonstrating the patency of third ventriculostomy fenestration. The limitations of MR imaging are that children often require a general anesthetic and that programmable shunt valves require reprogramming after MR imaging. In patients with acute hydrocephalus characterized by ventricular enlargement and transependymal edema with loss of sulci, the diagnosis is usually very obvious. However, there may be no reliable measurement values that can confirm or exclude the presence of hydrocephalus in a single series of images in some patients. Serial imaging demonstrating an increase in ventricular size may be required in equivocal cases. Conversely, an apparently normal ventricular size cannot exclude active hydrocephalus in a patient with a preexisting shunt, for example. A number of methods have been used to attempt to define hydrocephalus quantitatively on CT or MR imaging studies.72 Hydrostatic hydrocephalus is suggested in either of the following situations:
Both temporal horns have a width of 2 mm or more and the sylvian and interhemispheric fissures are not visible.
Both temporal horns have a width of 2 mm or more and the ratio of the largest width of the frontal horns to the internal diameter from inner table to inner table at this level is greater than 0.5.
Other features suggestive of hydrostatic hydrocephalus include the following: ballooning of the frontal horns of the lateral ventricles and third ventricle; periventricular hypoattenuation on CT or periventricular high-intensity signal on T2-weighted imaging and fluid-attenuated inversion recovery [FLAIR] sequences on MR imaging, suggesting transependymal exudate or migration of CSF; compression of the sulci and basal cisterns; upward bowing of the corpus callosum and downward displacement of the floor of the third ventricle on sagittal MR imaging. The Evans ratio is the ratio of largest width of the frontal horns to the maximal biparietal diameter. A ratio greater than 30 is suggestive of hydrostatic hydrocephalus. A modification of the Evans ratio, the frontal occipital horn ratio, may be more accurate and has been used in a number of prospective studies to quantify the degree of hydrocephalus and the response to treatment.73,74
9.8.4 Radiologic Criteria for Chronic Hydrocephalus
Skull radiographs may depict erosion of the sella turcica, or a “beaten copper” cranium. The latter can also be seen in craniosynostosis. The temporal horns may be less prominent than in acute hydrocephalus, and the third ventricle may herniate into the sella turcica. The corpus callosum may be atrophic; this is best appreciated on sagittal MR imaging. In infants with chronic hydrocephalus, sutural diastasis and delayed closure of the fontanels may be seen.
In communicating hydrocephalus, all ventricles are dilated. If the lateral and third ventricles are dilated and the fourth ventricle is small, it is likely that the obstruction is at the level of the aqueduct of Sylvius. MR imaging will help determine the cause, such as defining the presence of an obstructing tumor.
9.9 Diagnosis
Modern ultrasonography and, since the late 1980s, fetal MR imaging have significantly improved the ability to detect ventricular enlargement as a result of hydrocephalus in utero. Antenatal ultrasound and MR imaging provide reasonably detailed fetal brain anatomy, detect malformations, and can detect fetal ventriculomegaly as early as 17 to 21 weeks and 8 to 21 weeks, respectively.75,76 Normative data for ventricular size allow serial investigation during gestation.77
Anatomical ventriculomegaly is not sufficient to diagnose hydrocephalus. When making a diagnosis of hydrocephalus in neonates or infants, it is essential to establish that there is a truly abnormal rate of skull growth. Recording head circumference and comparing it with body weight and length centile charts are an integral part of the postnatal follow-up of any child. The head circumference must be recorded and plotted on an accepted growth curve chart with the patient’s exact age or, in premature infants, the gestational age. In the presence of hydrocephalus, any of the following may be observed: head circumference greater than 2 standard deviations above normal or out of proportion to body length or weight, upward deviations (crossing centile curves), or continued head growth of greater than 1.25 cm per week.
Evaluation of the patient with an enlarged head entails consideration of the many causes of macrocephaly, including hydrocephalus. Evaluation should include a history of trauma or CNS infection. The family history may demonstrate X-linked hydrocephalus caused by stenosis of the aqueduct of Sylvius or may reveal familial macrocephaly.
After a full review of the pregnancy, delivery, and neonatal history, as well as the clinical examination and ultrasound examination, it is usually possible to classify the hydrocephalus into an etiologic group. If no obvious explanation for the hydrocephalus can be found, then intrauterine infection should be investigated. Coagulation factor deficiency as well as thrombocytopenia should be excluded; isoimmune thrombocytopenia and coagulation factor V deficiency can present as congenital hydrocephalus resulting from congenital IVH.
9.9.1 Differential Diagnosis
Besides hydrocephalus, causes of increasing head size include chronic subdural effusion or hematoma, pseudotumor cerebri, neurofibromatosis, metabolic abnormalities of bone or brain, and cerebral gigantism (Soto syndrome). There are also benign familial forms.
Benign extracranial hydrocephalus is a condition of infants and children in which enlarged subarachnoid spaces are accompanied by increasing head circumference with normal or mildly dilated ventricles. This condition is also known as benign subdural collections of infancy or pericerebral CSF collections. It has been postulated by some to be a variant of communicating hydrocephalus but tends to run a benign course and stabilize by 12 to 18 months of age.78–80 Close serial monitoring of the head circumference and follow-up imaging with CT or MR imaging is recommended to monitor for ventriculomegaly. Shunting is rarely, if ever, required. A rare but striking condition that can mimic hydrocephalus is hydranencephaly, a post-neurulation defect that results in total or nearly total absence of the cerebral tissue; the intracranial cavity is filled with CSF. This is usually due to bilateral internal carotid artery infarcts or infection.81 Other brain malformations, such as agenesis of the corpus callosum and alobar holoprosencephaly, may also be associated with hydrocephalus but more often represent expansion of the third ventricle and separation of the lateral ventricles. Hydrocephalus ex vacuo is due to atrophy rather than altered CSF dynamics. Certain metabolic and degenerative disorders, such as glycogen storage and Alexander disease, can cause macrocephaly. Brain tumors of infancy may reach an enormous size, producing a large head apart from whether there is associated hydrocephalus. Finally, there may be a family history of large heads.
9.10 Pathology
The precise pathologic features of hydrocephalus vary depending on the age at onset, the rate of ventricular enlargement, and the degree of ventriculomegaly. Typically, elevated CSF pressure initially enlarges the frontal horns of the lateral ventricles, followed by enlargement of the entire ventricular system above the site of obstruction. Hydrocephalus is associated with flattening and destruction of the ventricular ependymal lining as well as edema and necrosis of the periventricular white matter.82 Periventricular glial cells proliferate, resulting in a layer of reactive gliosis. The pathologic findings may be a result of reduction in blood flow to the white matter, causing hypoxic injury, and/or toxicity to the white matter due to the buildup of waste products not removed appropriately because of changes in the extracellular matrix.83
In post-hemorrhagic hydrocephalus, high concentrations of proinflammatory cytokines,84 free iron, and hypoxanthine,85,86 which can generate highly reactive radicals, have been measured in the CSF.
Separation of the ependymal lining of the ventricles enhances permeability, which increases the edema of adjacent white matter (transependymal fluid absorption). The expanding ventricles flatten the cerebral gyri and obliterate the sulci over the cortical surface. Unless the acute obstruction is relieved, the increasing pressure may hinder cerebral blood flow, cause cerebral herniation, and compromise brainstem function.
The increasing pressure and ventricular enlargement are associated with necrosis of the brain parenchyma. White matter is more vulnerable to destruction than cerebral gray matter in the presence of progressive hydrocephalus.87,88 The corpus callosum may also be preferentially affected, with evidence of transcallosal swelling, thickening, or demyelination.89,90 These effects do not appear to be associated with cognitive changes or neuropsychological evidence of callosal disconnection.
9.11 Management
The management of hydrocephalus is the most common problem in pediatric neurosurgery. In infants and children with symptomatic or progressive ventriculomegaly, the decision to treat with a CSF diversion procedure poses no therapeutic dilemma. However, not all patients with enlarged ventricles require treatment.
In patients with obstructive hydrocephalus secondary to a mass that is surgically accessible, resection of the mass may lead to resolution of the hydrocephalus, and a shunt may not be necessary. This situation occurs infrequently in comparison with communicating hydrocephalus. If no documented obstruction or operable lesion is present and the hydrocephalus is slight and slowly progressive, a trial period of observation or medical management may be indicated, especially in preterm infants.
Another situation in which observation is reasonable is arrested hydrocephalus, which is an uncommon state of chronic hydrocephalus in which the CSF pressure has returned to normal and there is no pressure gradient between the cerebral ventricles and the brain parenchyma. Patients should be followed carefully with neurologic examinations, neuropsychological assessments, and careful assessment of their development. A shunt will be necessary if there is any deterioration of those parameters.
Rapid-onset hydrocephalus with increased ICP is an emergency. Depending on the specific case, any of the following procedures can be performed: ventricular tap in infants; external ventricular drainage and lumbar puncture in post-hemorrhagic and post-meningitic hydrocephalus; endoscopic third ventriculostomy; and ventriculoperitoneal shunt.
The management of patients with in utero hydrocephalus remains controversial. Although the results of surgical attempts in the 1980s were seen to be ineffective, interest remains as diagnostic technology and fetal surgery techniques advance.91 The recent Management of Myelomeningocele Study (MOMS), in which patients with a fetal diagnosis of myelomeningocele were randomized to either in utero closure or expectant treatment and postnatal closure, showed a significant increase in the requirement for CSF diversion, from 98 to 68%, albeit with the increased risk for preterm labor and maternal complications.92
9.12 Prognosis
Before the 1950s, the outlook for patients with untreated hydrocephalus was extremely poor. Forty-nine percent of patients had died by the end of the 20-year observation period, and only 38% of survivors had an IQ greater than 85.93 The development of satisfactory shunting substantially improved the outlook for patients with hydrocephalus but brought its own set of problems and complications. Most children with hydrocephalus require multiple shunt revisions. Shunt dependence carries with it an annual mortality rate of 1%.94,95 Another series, with 907 patients, reported a mortality rate of 12% at 10 years,96 with the main risk factor for death being a history of shunt infection.
Shunt-related complications, including death, have been reported to be greater in patients with myelomeningocele than in those who required shunt placement for the treatment of other conditions.39,94
The neurologic and intellectual disabilities of patients with hydrocephalus depend on many factors, including the etiology and severity of the hydrocephalus, thickness of the cortical mantle and corpus callosum,97 requirement for a shunt, and presence of other brain anomalies.98 Associated conditions, such as IVH, CNS infection,96 and hypoxia, may dictate the ultimate prognosis more than the hydrocephalus.
A series of 233 patients with congenital hydrocephalus were evaluated for longer than 20 years; 13.7% died. The average number of shunt revisions was 2.7. In this series of 233 patients, of 115 who underwent psychological evaluation, approximately 63% showed normal performance, whereas 30% had mild retardation and 7% had severe retardation.99 Another study found that children with congenital hydrocephalus were less likely to require special education placement (29%) than were those in whom hydrocephalus was due to meningitis (52%) or IVH (60%).100
Epilepsy also is more prevalent among patients with hydrocephalus, and complications of shunt surgery appear to play a relatively minor role in its development.101
Intellectual sequelae include significant scatter among Wechsler Intelligence Scale for Children-Revised (WISC-R) subtest scores, often with greater impairment of performance and motor tasks, as well as of nonverbal compared with verbal skills.102 Normal intellectual function is present in 40 to 65% of patients who receive appropriate treatment.98 The probability of normal intelligence is enhanced if shunts are placed early and proper function is maintained. A study of 99 children ranging in age from 6 to 13 years with shunted or arrested hydrocephalus demonstrated a close correlation between area of the corpus callosum affected and impairment of nonverbal cognitive skills and motor abilities.103 Behavioral problems also are more common in children with hydrocephalus, irrespective of etiology.104
9.13 Conclusion
Pediatric hydrocephalus is a common, complex, and in many ways poorly understood disorder. Although the persistent efforts to improve upon the treatments developed over the last century have had modest success, recent advances in imaging, neurophysiology, and molecular biology have led to important discoveries, suggesting that significant advances are imminent.
Pearls
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


