Figure 87.1. Preliminary cellular model for HH epileptogenesis based upon laboratory findings derived from surgically resected HH tissue [references (21–27); reviewed in (22)]. A photomicrograph of HH tissue is shown on the left side of the figure (hematoxylin and eosin stain). A small HH neuron (typically <16 μm in diameter) is indicated by the arrow, characterized by the well-defined nuclear membrane and densely staining nucleolus (21). A cluster of small neurons is seen immediately to the right of the arrow. The working model for epileptogenesis is shown on the right side of the figure. Small HH neurons tend to occur in clusters (21). They also express GAD and demonstrate intrinsic, spontaneous pacemaker-like firing activity in microelectrode recordings from freshly resected HH slice preparations (23). Their projections appear to be local, indicated by the solid line connecting two small HH neurons (24). Large HH neurons (typically >20 μm in diameter) have the morphology of projection neurons and may express excitatory neurotransmitters (24). These large neurons depolarize to GABA agonist administration (26,27). The evidence for structural and functional connections between small and large HH neurons is incomplete, indicated by the use of a dashed line. Small HH neurons within the cluster may be functionally connected by gap junctions (unpublished observation). The destination of the axonal projections of large HH neurons is unknown, also indicated by dashed lines. We hypothesize that clusters of small spontaneously firing HH neurons are linked in a functional network, resulting in the synchronized release of GABA, which has an excitatory effect on the larger projection neurons (22). (Copyright Barrow Neurological Institute, 2013.)
ETIOLOGY
Most HH cases are sporadic, and the underlying cause is unknown. However, HH is a cardinal feature of Pallister–Hall syndrome, known to result from haploinsufficiency of GLI3, a zinc finger transcription factor in the sonic hedgehog pathway. Somatic mutations (mutations present in the tumor only) in GLI3 are associated with HH in approximately 15% to 25% of sporadic cases, based upon current genotyping technology (28,29).
While somatic mutations in GLI3 may be responsible for sporadic HH in some cases, other mutations are also likely to be discovered. At least two other susceptibility loci have been reported, specifically SOX2 and 6p25.1-25.3, a locus which includes FOXC1 (30,31). Like GLI3, SOX2 and FOXC1 are known to be transcription factors that are active during morphogenesis of the ventral forebrain. A project utilizing whole exome sequencing of DNA derived from HH compared to whole blood is currently under way (unpublished observation).
The specific molecular mechanisms controlling cellular proliferation that result in HH are unknown.
CLINICAL FEATURES
There is a great deal of diversity with respect to the age of onset, severity, and evolution of the neurologic symptoms in patients with HH and epilepsy (32). The tremendous clinical variability from case to case must be kept in mind when evaluating patients with a possible diagnosis of HH. These same clinical features, particularly the presence or absence of neurologic deterioration and the pace at which it is occurring, affect the decision-making process for deciding the type and timing of therapeutic intervention.
Epilepsy: Gelastic Seizures
Gelastic seizures are the most specific symptom associated with HH. They are usually brief, typically just a few seconds in duration, and usually last <30 seconds. They usually occur daily, with multiple seizures per hour in more severely affected patients. Gelastic seizures can be associated with little or no change in consciousness, particularly early in the clinical course, although making this determination in infants and young children can be challenging. Superficially resembling laughter, the patients generally do not experience mirth, and most family members can readily distinguish the gelastic seizure from true laughter. Not uncommonly, patients may have clinical events that more closely resemble crying rather than laughing (ictal crying or dacrystic seizures). Gelastic seizures can also be quite subtle. A purely subjective sensation, described as a pressure to laugh, can be described by communicative patients (33). They are commonly mistaken for other conditions, particularly during early infancy, including colic and gastroesophageal reflux disease (34).
Gelastic seizures associated with HH begin at an early age and are usually the first seizure type (35). In our experience, the clinical diagnosis is almost always delayed by months or even years. In retrospect, parents can identify the onset of peculiar laughing spells at a very early age. The mean age of onset for gelastic seizures in our series of HH patients with refractory epilepsy (N > 180) is 11 months, with onset before 1 month of age in 45% and before 1 year of age in 81%. However, in this series, seizures have presented as late as age 13 years, emphasizing the clinical diversity within the HH population. Gelastic seizures become less frequent during the first decade and may disappear entirely as other seizure types develop (36). Uncommonly, patients with HH may not develop gelastic seizures until early adulthood (37).
The EEG features associated with HH and gelastic seizures deserve emphasis, specifically because ictal recordings, obtained with the conventional placement of electrodes over the scalp, often show no change in the EEG from the ongoing background, which itself is often normal (38–40). Hence, clinicians need to be alert to this fact so as to not miss the correct diagnosis of epileptic seizures. Alternatively, nonlocalizing ictal changes may be observed, such as relative flattening of the EEG background, generalized paroxysmal fast activity, or an absence of interictal spikes (4,32,41,42). There is limited utility to video–EEG seizure monitoring for most patients with HH and epilepsy (42).
Gelastic seizures do not usually respond to antiepilepsy drugs (AEDs). Consequently, the timing of surgical intervention (here, this term includes GK radiosurgery) is the major decision point facing the patient, family, and clinician. Brief, infrequent gelastic seizures are not disabling. If the child is making good developmental progress, a decision to withhold surgical intervention is appropriate. However, under these circumstances, the clinical course needs to be observed carefully for any adverse changes.
Epilepsy: Other Seizure Types
Seventy-five percent of patients with gelastic seizures and HH will develop other types of seizures (35,36). The age at which other seizure types will appear varies, but is most likely to occur between 4 and 10 years of age (4). Mullati et al. (36) have reported two to five seizure types for each patient with childhood onset of epilepsy due to HH, and virtually all seizure types have been reported, including infantile spasms accompanied by hypsarrhythmia (43). A review of the published reports regarding lifetime prevalence of seizure types in patients with HH suggests that complex partial seizures occur in 50% to 60% of patients, tonic–clonic seizures in 40% to 60%, atypical absence in 40% to 50%, tonic seizures in 15% to 35%, and “drop attacks” in 30% to 50% (32,35,36,44–46). Seizures associated with HH are usually refractory to management with AEDs (47).
When they occur, complex partial seizures often suggest temporal lobe localization (most frequently) or frontal lobe localization based upon seizure semiology and the results from conventional video–EEG seizure monitoring utilizing scalp electrodes. However, surgical outcomes following temporal lobe or frontal lobe resections in HH patients are universally poor (48).
Freeman et al. (49) have reported the presence of a symptomatic generalized epilepsy phenotype in 12 of 20 patients undergoing HH resection. Their cohort of patients demonstrated features of Lennox–Gastaut syndrome, including tonic seizures, and slow spike-wave and polyspike activities on interictal EEG. Seizure onset (gelastic seizures were the first seizure type in 92% of these patients) began between birth and 24 months of age (mean 0.3 years), while tonic seizures developed between 2 months and 9 years of age (mean 6 years).
The interictal EEG is frequently normal early in the natural history of epilepsy associated with HH, particularly when gelastic seizures may be the only seizure type (32,35,36,42). However, the appearance and subsequent evolution of abnormal EEG findings parallel the worsening of the epilepsy with the emergence of multiple seizure types (4,35,36,49). In the review of Tassinari et al. (35), EEG studies in HH patients with multiple seizure types showed normal results in only 2%, generalized spike or spike-wave findings in 47%, multifocal independent spikes in 18%, and focal spikes (most frequently over the temporal regions) in 33%. Localization of the epileptic process in HH is complex, as seizures (simple partial, complex partial, or secondarily generalized) can originate within the HH and spread to cortical regions. These seizures may or may not have a clinically apparent gelastic component at the onset.
However, the observed changes in seizure type and the evolution of EEG findings also suggest a process of secondary epileptogenesis, in which distant cortical structures begin to generate seizure events that are independent of the original seizure focus (in this case, the HH lesion) (50–53). Initially, the new focus is dependent upon the presence of the original focus, such that removal of the HH will lead to a decrease in seizure frequency and eventually complete disappearance of seizures arising from the second focus (the “running-down phenomenon”) (51). With time, however, usually over a period of years, the second focus becomes entirely independent of the original, inciting focus, such that its removal does not influence the independent epileptogenesis of the second focus. The original concept of secondary epileptogenesis was formulated in the context of temporal lobe epilepsy (54), but epilepsy associated with HH is also consistent with this model (46).
Video–EEG recordings with intracranial electrode implantation have demonstrated that seizure activity developing later in the course of the disease may not arise from the HH lesion (5,49,50,55). Approximately 10% of patients undergoing resective surgery for HH experience the running-down phenomenon, in which seizures of neocortical origin decrease in frequency and eventually stop over a period of weeks or months (56). The running-down phenomenon is observed in 21% to 40% of those HH patients who are ultimately seizure free following surgical resection (at least 1 year of postoperative follow-up) (56,57). Conversely, failure of surgical treatment in HH cases may be attributed to secondary epileptogenesis, as patients with 100% HH lesion resection may continue to have residual seizures in the absence of any other identifiable structural lesion (56).
Although the possibility that there are other cerebral abnormalities must be considered (58,59), most HH patients do not have observable structural abnormalities by high-resolution magnetic resonance imaging (MRI) (56,60–63). The cellular mechanisms for secondary epileptogenesis and the running-down phenomenon are unknown (53).
Cognition and Development
The clinical course of worsening epilepsy and increasingly abnormal EEG findings can also be accompanied by developmental regression and cognitive decline (4,32,64–67). There is a great deal of individual variability in this regard, but approximately 50% of HH patients with the onset of seizures during infancy will experience this deteriorating clinical course (47). Therefore, the degree of impairment demonstrated by any individual patient is a potentially moving target. There are no published series that document this natural history with longitudinal study of a large cohort of patients, but those detailed individual case reports that are available are compelling (64,68).
Cognitive impairment is common in HH patients, with or without the deterioration noted above, occurring in 80% or more of the patients with the intrahypothalamic subtype of HH (35,44,69). Cognitive problems correlate with the presence of epilepsy as a comorbid feature. Patients with parahypothalamic HH typically do not have epilepsy, and also have little or no cognitive impairment (70). The severity of cognitive impairment and developmental retardation correlates with an earlier age of seizure onset (47), and HH lesion size and subtype (69).
Behavior and Psychiatric Symptoms
Patients with epilepsy and intrahypothalamic HH lesions also have a high likelihood to develop clinically significant behavioral and psychiatric problems (4,44,71). These symptoms can present the most significant day-to-day problem for affected families and in some instances lead to placement outside the home. Mood lability and rage attacks are the most frequent symptoms. Patients can have poor frustration tolerance, with acting-out behavior and excessive reactivity to relatively minor stimuli, sometimes with destructive and aggressive features (72).
There is a strong positive association between the incidence of refractory epilepsy, cognitive impairment, and behavioral disturbance in HH patients (47). There is abundant descriptive literature that worsening seizures, cognitive decline, and behavioral deterioration occur simultaneously (4,64,65,73,74). HH is a clinical model for epileptic encephalopathy, although the basic mechanisms responsible for this are unknown.
TREATMENT
Rationale for the HH as the Therapeutic Target
There is now compelling evidence that gelastic seizures arise from HH tissue (66). This idea was slow to gain acceptance, since localization-related seizures were thought to arise exclusively from cortical structures (75,76). However, Kahane et al. (5,55) reported in 1994 that if ictal video–EEG recordings included intracranial monitoring with an electrode in the HH, then the ictal EEG pattern associated with gelastic seizures was initially seen in the HH lesion. This has subsequently been confirmed by multiple additional reports (50,71,77–79). Electrical stimulation of the electrode contacts within the HH can provoke the patient’s habitual gelastic seizures (50,77,79). Functional imaging with single photon emission computed tomography (SPECT) has demonstrated increased perfusion in the HH with ictal SPECT imaging (77,80), and ictal imaging with flourodeoxyglucose positron emission tomography has also shown increased metabolism within the HH lesion (59,81). Perhaps, the most important evidences for the intrinsic epileptogenesis of HH lesions are the outcomes observed with surgical resection, in which gelastic seizures can be abolished with successful surgical removal or disconnection.
Absence of Controlled Treatment Trials
There are no controlled trials investigating treatment issues for HH and epilepsy. However, over the past decade, multiple uncontrolled treatment series have been published as HH cases became concentrated at multidisciplinary referral centers (56,57,61,63,82–89).
Antiepilepsy Drugs
There is broad consensus in the literature about the lack of efficacy of AEDs (47). It is likely that the number of medication-responsive patients is underestimated due to the ascertainment bias of epilepsy referral centers, and a small number of cases responsive to AEDs have been reported (8,33,81). However, probably <5% of patients with intrahypothalamic HH and epilepsy achieve complete and sustained seizure control with medications alone. AEDs are often reported to have little impact on the frequency of gelastic seizures but may be valuable by reducing the frequency of other seizure types (66). At this time, no AED has emerged as demonstrating superior efficacy for treating epilepsy associated with HH. As a consequence, AEDs are probably best chosen based upon other factors, including adverse event profile and ease of administration.
Presurgical Evaluation
Video–EEG seizure monitoring is a conventional component of the evaluation process for epilepsy surgery. However, as the HH lesion is deep in the brain, the results of seizure monitoring with electrode placement over the scalp have limited utility, and these results should be used with caution when planning surgical interventions (42). In general, in HH cases, seizure monitoring is more likely to identify patterns of ictal spread, rather than localizing seizure onset. Even for those patients with secondary epileptogenesis, seizure activity arising from the second focus is dependent (for an undetermined time) upon the presence of the HH (see the discussion of the running-down phenomenon). Accordingly, for almost all patients with HH and epilepsy, the HH lesion is the most appropriate initial surgical target.
Invasive seizure monitoring, with electrodes implanted into the HH as well as multiple other brain regions, was necessary to prove seizure localization in HH cases. However, this type of implantation is technically challenging, has a low but definite risk of surgical complications, and rarely alters the decision-making process. Accordingly, we do not recommend intracranial monitoring for most HH patients.
The timing of surgical intervention is influenced by the emergence of multiple seizures types, often accompanied by cognitive and behavioral regression. Neuropsychological testing is recommended at yearly intervals, if possible, to monitor for changes that may not be immediately apparent in the classroom or in the home.
MRI is the most important modality for diagnosis and surgical planning. For HH patients, MRI should include a coronal T2 fast spin-echo (FSE) sequence with thin cuts and no imaging gaps through the hypothalamus. The presence or absence of structural abnormalities elsewhere in the brain must also be determined.
Surgical Resection/Disconnection
Patients with the parahypothalamic (or pedunculated) subtype of HH usually do not have epilepsy or cognitive disturbance and are medically treated with gonadotropin-releasing hormone agonists (such as leuprolide acetate). Accordingly, surgical resection is usually not indicated for this subgroup of HH patients (8,90).
However, some of the early case reports of resective surgery for patients with CPP secondary to HH happened to include children with gelastic seizures, and improvement in seizure control was noted (91,92). Subsequent reports with HH resection specifically for epilepsy indicated encouraging results for seizure control in some patients and suggested an improved outcome for cognitive and behavioral functioning (67,93,94).
The last decade has seen significant improvements in the operative techniques available for surgical resection and/or disconnection of HH lesions associated with epilepsy. The relative merits of one treatment approach over another are based upon the individual circumstances of each patient, including the surgical anatomy of the HH. The clinical course for each patient, particularly as it relates to any signs of regression or worsening, also influences the decision to use one treatment modality over another, as well as the timing of intervention.
HH Classification and Surgical Anatomy
When considering the surgical options, the classification of HH lesions must be refined beyond the binary model used thus far, specifically, intrahypothalamic and parahypothalamic HH subtypes. Several authors have proposed classification schemes for HH lesions, including Valdueza et al. (8), Regis et al. (62), and Delalande and Fohlen (82,95). Regardless of the classification system that is used, our experience suggests that there is a relatively smooth continuum between these subtypes. However, while the advantages of one classification scheme over another are debatable, each of these schemes addresses an important issue: the surgical anatomy of the HH lesion. Currently, our preference is to utilize the Delalande classification system (82,95) (Fig. 87.2).
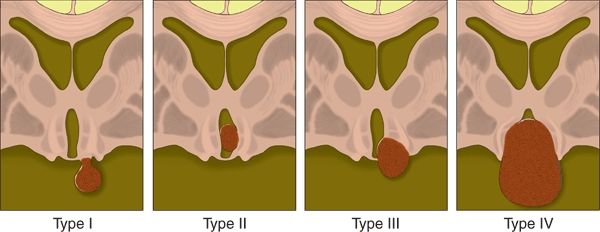
Figure 87.2. Classification system for HH, proposed by Delalande and Fohlen (95). Type I lesions have a horizontal base of attachment, below the normal position of the floor of the third ventricle. If attached by a narrow stalk to the tuber cinereum, they result in CPP, whereas more posterior attachment to the region of the mammillary bodies may result in epilepsy. Larger type I lesions can result in both CPP and epilepsy. Type II lesions have a vertical plane of attachment to the wall of the third ventricle, completely above the normal position of the floor of the third ventricle. Type III lesions may be unilateral or bilateral and have a plane of attachment that extends both above and below the floor of the third ventricle. Consequently, these lesions have both vertical and horizontal planes of attachment when viewed on a coronal sequence. Type IV lesions are termed “giant,” though features clearly distinguishing them from type III lesions were not provided (95). (Copyright Barrow Neurological Institute, 2013.)
Selecting the optimal surgical approach must take into account the location and size of the HH lesion, and most importantly, the anatomy of its base of attachment to the hypothalamus (82,95–98). Type I lesions in the Delalande system are attached to the inferior (horizontal) surface of the hypothalamus. This type includes those HH lesions with a thin peduncle or stalk, often attached to the tuber cinereum, and associated with only with CPP, but can also include HH lesions with a broader or more posterior base of attachment that are associated with epilepsy. These lesions are best resected or disconnected by an inferior or pterional approach. Conversely, Delalande type II lesions have a vertical plane of attachment within the third ventricle and are best suited to a superior surgical approach to resection (transcallosal interforniceal or transventricular endoscopic [TE]), stereotactic thermal ablation, or GK radiosurgery. Delalande types III and IV have both vertical and horizontal planes of attachment (both above and below the normal position of the floor of the third ventricle). The superior approaches noted above may be adequate, but some of these cases may require a combined approach, with either simultaneous or staged resections.
Pterional Approach
Until 2001, resective surgery for HH was almost always performed with a surgical approach from below the lesion. As reported by Nishio et al. (94,99) and Machado et al. (67) in detailed case studies, resection of the HH via a pterional approach had the potential to control seizures and improve the patient’s cognitive and behavioral level of functioning. Other surgical approaches for the HH lesion from below have also been reported, including orbitozygomatic (98), subfrontal (71), and lamina terminalis approaches (71). In those instances where a complete resection via a pterional approach is possible, seizure outcomes are good (66% seizure free with complete resection of the lesion) (47,86). Pterional resection (in our hands usually an orbitozygomatic approach) is still the most appropriate means of resection for HH cases with epilepsy and type I lesions (86).
However, the pterional approach is not suited to the surgical anatomy of most HH cases, where a substantial component of the HH has a vertical plane of attachment within the third ventricle (Delalande types II to IV). Additionally, the complication rate, including stroke and cranial nerve injury, was substantial in earlier series (47,71). These approaches traverse territory with vascular structures, including the internal carotid artery, anterior and posterior communicating arteries, and their associated perforating branches. The optic tracts and chiasm and the third cranial nerve are also vulnerable (96).
Transcallosal Anterior Interforniceal (TAIF) Approach
Although utilized previously for other pathologies (100), Rosenfeld et al. (6,97) in Melbourne, Australia, were the first to utilize the TAIF approach to the third ventricle to resect HH lesions in patients with refractory epilepsy. This approach, utilizing microsurgical technique and intracranial guidance systems, allows for excellent direct visualization of the HH and its base of attachment within the third ventricle. Rosenfeld’s modification of the transcallosal approach to the third ventricle, with a more anterior, transseptal trajectory, minimizes retraction of the columns of the fornix and also avoids injury to the internal cerebral veins, located more posteriorly (97).
The Melbourne group has published a series of 29 consecutive patients undergoing intrahypothalamic HH resection via the TAIF approach (57). Age at surgery ranged from 4 to 23 years (mean age 10 years). All patients had multiple seizure types, including gelastic seizures. Coexisting morbidities included a history of CPP in 13 (45%), intellectual disability in 21 (72%), and behavioral problems, most frequently rage and aggression, in 18 (62%).
At least 95% resection of HH lesion volume was achieved in 18 patients (62%). Postoperative follow-up for a minimum of 12 months showed 15 patients (52%) who were completely seizure free and 7 patients (24%) with at least a 90% improvement in seizure frequency. Surgical resection was generally well tolerated. Small, unilateral ischemic strokes of the thalamus and internal capsule occurred in two cases (7%), both with complete recovery, and transient third cranial nerve injury was reported in one patient. The majority of patients (55%) developed mild, asymptomatic hypernatremia postoperatively, but no patients had persistent disturbances in fluid or electrolyte homeostasis. Five patients (17%) required thyroid hormone replacement therapy following surgery. Increased appetite with weight gain was reported in 45% of patients but resolved in half of these with time.
Impairment of short-term memory was, however, a significant issue. The TAIF surgical approach, despite its more anterior trajectory, requires retraction of the columns of the fornix. Transient memory disturbance was noted in 14 patients (48%) during the immediate postoperative period, but residual difficulties were reported by only 4 (14%). Attention and behavior were noted to improve in many of the patients in this series, but further details were not available (57).
Very similar results were subsequently reported by Ng et al. (56) at the Barrow Neurological Institute in Phoenix. In this series of 26 consecutive patients undergoing TAIF, 54% were completely seizure free. The likelihood of complete seizure freedom (with at least 1 year of postoperative follow-up) had a positive correlation with the percentage of HH lesion volume that was successfully resected (P < 0.05). The risk and type of surgical complications were also similar. Notably, transient short-term memory impairment was noted in 58% of the patients but persisted in only two patients (8%).
Transventricular Endoscopic Approach
Transcortical TE resection and/or disconnection is also a treatment option for HH patients with refractory epilepsy (61,71,82,85,95,101–104). Barrow has reported a series of 37 consecutive HH patients treated with endoscopic resection/disconnection for treatment-resistant seizures (61). All patients had at least 1 year of follow-up. The mean age at the time of surgery was 11.8 years (range 0.7 to 55 years). All patients had a history of gelastic seizures at some time point during their clinical course, and 29 (78%) had active gelastic seizures at the time of surgery. Twenty-eight patients (76%) had type II HH lesions by the Delalande classification, and the median lesion volume was 1.0 cm3. (In comparison, in the Barrow series of transcallosal resections noted above, 42% had a type II lesion, and the median HH lesion volume was 2.4 cm3, reflecting the different selection criteria for each approach (56).)
Median follow-up was at 21 months. Eighteen patients (49%) were completely seizure free, while seizure frequency was reduced at least 90% in an additional eight patients (22%). Twelve patients were determined to have 100% of their HH lesions resected. Of these, eight (67%) were 100% seizure free (61).
As observed with the TAIF approach, most patients tolerated endoscopic resection well. However, some differences from the TAIF approach were observed, with a significantly shorter total length of hospital stay in the endoscopic group (mean 4.1 days) versus the previously reported transcallosal group (mean 7.7 days, P < 0.001) (56,61). Only five patients (14%) experienced postoperative short-term memory loss, but this appeared to be a permanent residual problem (by history) in three (8%), which is comparable to the TAIF approach. No patients with endocrine disturbance, either transient or permanent, were observed. However, eleven patients (30%) showed small unilateral thalamic infarcts on diffusion-weighted MRI sequences. These were clinically asymptomatic in nine of eleven cases and the remaining two patients made a complete clinical recovery. These infarcts were attributed to disruption or injury to small thalamic perforators as a result of local brain movement with excursions of the endoscope.
Wethe et al. (105) at Barrow have recently reported the postoperative neuropsychological testing results for patients with HH and treatment-resistant epilepsy. Of the cohort of 32 patients, 63% underwent endoscopic resection. For the entire cohort, there was a statistically significant improvement in full-scale intelligence quotient (IQ), with a preoperative mean of 83.0 and postoperative mean of 91.3 (P < 0.001). For the entire cohort, there was no significant difference for preoperative and postoperative scores relating to learning and memory. Improvement in cognitive functioning was most likely to occur in patients who were younger at the time of surgery (and had a shorter lifetime duration of epilepsy) and in those with lower scores with preoperative testing (105).
For those HH patients who require surgical resection/disconnection from above, the factors that favor the endoscopic approach include smaller lesions, unilateral attachment, adequate space within the third ventricle to manipulate the endoscope, and adequate size of the lateral ventricle and foramina of Monro for safe instrumentation. Factors that are more favorable for the TAIF approach include a younger age at the time of surgery (the columns of the fornix and leaves of the septum tend to fuse with age), larger lesions, and bilateral attachment.
Gamma Knife Radiosurgery
GK radiosurgery is now established as an ablative therapy for patients with HH and treatment-resistant epilepsy (55,83,101,106–113). GK is noninvasive and can deliver a clinically effective dose of radiation to a small volume of tissue via a large number of independent trajectories, with little or no injury to surrounding brain.
Regis et al. (114) have described a series of 27 patients with intractable epilepsy and HH with at least 3 years of follow-up after GK therapy. GK delivers its maximal destructive energy to the interior of the targeted lesion, and the intensity of energy delivery falls off toward the periphery of the lesion. A dose of at least 17 Gy is ideally delivered to the entire lesion. The peripheral treatment margin (usually referred to as the 50% isodose margin) is matched to the outer edge of the HH lesion, but the dosimetry map may need to be modified in proximity to the optic tracts or other radiosensitive structures.
A maximal threshold of 10 Gy to the optic tracts and 8 Gy to the optic chiasm and optic nerve was utilized for treatment planning for this prospective treatment study (114). In this series, the median HH diameter was 0.95 cm (range 0.5 to 2.6 cm) and the median volume of the marginal isodose was 0.65 cm3 (range 0.13 to 2.67 cm3). The median radiosurgery dose to the 50% isodose margin was 17 Gy (range 13 to 26 Gy, mean 16.9 Gy). Of the 27 patients reported, 10 (37%) were completely seizure free and an additional 6 (22%) were substantially improved with only rare gelastic seizures.
Efficacy is delayed from the time of GK treatment. Initially after treatment, seizure frequency may be improved, or patients may continue to have seizures at their pretreatment baseline. Several months following therapy, an increase in seizure frequency, lasting for only a few days up to several weeks, may be observed. Subsequent to this, patients responding to treatment will experience progressively fewer seizures, with complete seizure control after a period of 6 to 24 months. Regis et al. recommend waiting 36 months from the time of treatment to assess final efficacy.
GK has an excellent adverse event profile. Most patients have no complications or side effects attributable to GK treatment. No patients among the 27 treated were reported to have a permanent complication (114). Three patients (11%) experienced transient poikilothermia. In contrast to side effects that may be seen with resective surgery, there were no patients in this series that experienced weight gain, endocrine disturbance, adverse changes in cognition, or short-term memory complaints. The disadvantage of GK is the delayed onset of action for controlling seizures and the more limited anatomical spectrum of HH lesions to which it is suited.
GK is an important treatment option for many patients with HH and epilepsy, and should be the preferred treatment modality for smaller lesions, particularly for patients who are clinically stable and capable of tolerating the delay in efficacy to obtain improved seizure control. It is a less desirable approach for those patients who are progressively worsening with their epilepsy or experiencing cognitive decline or behavioral deterioration with uncontrolled seizures. Additional study to define the optimal role of GK compared to other treatment modalities is required.
Stereotactic Thermal Ablation
Radiofrequency thermal ablation has been described in a relatively small number of patients (37,77,79,87,115–118). This technique involves stereotactic placement of a depth wire into the HH target and then causing a destructive thermal lesion by physically heating the probe tip. Most of these publications are single case reports.
Kameyama et al. (87) have reported a series of 25 HH patients undergoing stereotactic thermal ablation with a follow-up interval of at least 6 months. Mean age was 14.8 years (range 2 to 36 years) at time of treatment. All patients had at least daily gelastic seizures, and 22 (88%) had multiple seizure types. HH lesions were classified as intrahypothalamic in 10 (corresponding most closely to Delalande type II; 40% of treatment cohort), parahypothalamic in 6 (corresponding to Delalande type I; 24%), and mixed in 9 (corresponding to Delalande types III and IV; 36%).
The radiofrequency thermocoagulation probes were placed with MRI-guided stereotaxis and heated to 74°C for 60 seconds, resulting in a 5-mm spherical lesion. Targeting prioritized the HH lesion at the point of attachment, maximizing the potential for disconnection, along with the central region of larger lesions. Up to 4 lesions could be created by moving the probe within its track. Additional passes were required for most patients to ablate the intended target for each individual patient (mean lesions per patient 7.2 [range 1 to 18] and mean tracks per patient 3.8 [range 1 to 8]). Six patients (24%) required a second thermocoagulation procedure to obtain final results.
Complete freedom from seizures (Engel class I) is reported in 76%. Gelastic seizures disappeared in 92% of patients. Results were more favorable in the pediatric subgroup with complete seizure control in 16 of 18 patients (89% Engel I), whereas seizures were completely controlled in only 3 of 7 adults (43% Engel I). There were no permanent complications. Transient problems following thermocoagulation treatment included hyperthermia (n = 4 patients), hyperphagia (n = 2), hyponatremia (n = 4), Horner’s syndrome (n = 3), and short-term memory disturbance (n = 2). There was a significant increase in IQ or developmental quotient postoperatively (preoperative mean 67.6 and postoperative mean 75.9; P < 0.0001).
Stereotactic thermal ablation is emerging as the preferred treatment modality for many HH lesions, but expertise is available in a small number of centers. Further research is required to define the boundaries within which stereotactic thermal ablation is the optimal surgical approach versus microsurgical or endoscopic resection.
A recent technical innovation on the use of stereotactic thermocoagulation utilizes laser energy to heat the tip of the stereotactic probe and has the added advantage of near real-time MRI thermography to follow the delivery of the treatment dose with predetermined safety parameters (119). Curry et al. (120) have reported two HH patients treated with this device, both of whom were completely free of seizures with brief follow-up (5 and 2 months, respectively) at the time of initial publication. This technique (MR-guided laser interstitial thermal therapy) appears promising, but thus far, a limited number of patients with HH have been treated worldwide. Additional peer-reviewed research reports with a larger cohort and long-term follow-up are anticipated (120).
Interstitial Radiosurgery
Interstitial radiosurgery with stereotactic implantation of 125I radioactive seeds has also been proposed as an ablative therapy for HH associated with epilepsy (63,121). Schulze-Bonhage et al. (121) in Freiburg, Germany, have reported a series of 24 patients (mean age 21.9 years), all of whom had treatment-resistant gelastic seizures, in addition to other seizure types. Mean HH lesion volume was 1.2 cm3.
The treatment plan was designed to deliver a dose of 60 Gy at the outer margin of the HH, followed by radioisotope seed removal. Thirteen of twenty-four patients (54%) required at least one reimplantation for a second course of therapy if the response to the initial course was unsatisfactory. With follow-up of at least 2 years, 12.5% were seizure free (Engel I outcome) while 41.7% had at least a 90% improvement in seizure frequency (121). Treatment response is described as occurring within 8 weeks following treatment. No complications were noted, but follow-up MRI 3 months after treatment revealed local cerebral edema in five of twenty three patients (22%), in some instances associated with headache and fatigue. Neuropsychological testing prior to implantation and at least 1 year following treatment showed no significant changes with groupwise analysis of the treatment cohort.
Staged Procedures for Giant HH
For the purposes of this discussion, giant HH lesions are defined as exceeding a volume of 4 cm3, as determined by measuring the diameter of the lesion in the three major axes and applying the formula for determining the volume of an ellipsoid [vol = π × height × length × width/6]. HH lesions of this size account for 12% of our surgical series (22 of 180 patients). These lesions are problematic as patients with giant HH are at higher risk for severe epilepsy (122), developmental impairment (69), and CPP (13,60).
They are also challenging lesions with respect to surgical therapy, relating more to the nature of their attachment to the hypothalamus rather than to their size alone. Most giant HH lesions have both vertical (above the normal position of the floor of the third ventricle) and horizontal (below the normal position of the floor of the third ventricle) planes of attachment, and consequently, no one approach, from either above or below, may enable the surgeon to safely visualize the plane of attachment and adjacent structures (Fig. 87.3).
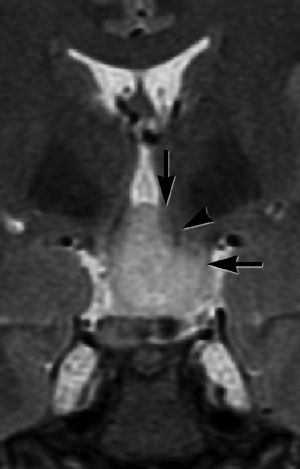
Figure 87.3. Delalande type IV (giant) HH, as shown on coronal T2-weighted FSE sequence, illustrating the complexity of attachment for these large lesions. There is a largely vertical plane of attachment within the third ventricle (vertical black arrow) and a largely horizontal plane of attachment below the hypothalamus (horizontal black arrow). Note the dark signal corresponding to the left descending column of the fornix (black arrowhead), which partially traverses the HH to lead to the mammillary body (located posterior to this slice). (Copyright Barrow Neurological Institute, 2013.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


