Inflammation
Annamaria Vezzani
Jukka Peltola
Damir Janigro
Introduction
Immune and inflammatory reactions have been described in the central nervous system (CNS) in various neurologic diseases, including epilepsy. In particular, several cytokines and activated proinflammatory signaling pathways have been measured in the brain, cerebrospinal fluid (CFS), or blood after seizures induced in experimental models and in clinical cases of epilepsy (for review see Vezzani101).
Inflammatory reactions in the CNS appear to arise from the activation of both branches of the immune system, namely, innate and adaptive immunity. In the rodent brain, inflammatory mediators are produced during epileptic activity by microglia, astrocytes, and neurons as part of innate immune mechanisms. In human epileptic tissue, evidence suggests that both innate and adaptive immunity contribute to the induction and perpetuation of inflammation in the brain.63 The blood–brain barrier (BBB) appears to play a crucial role in modulating the functional communication between innate and adaptive CNS immunity by tightly regulating the entry of blood-borne immune cells, antigens, and antibodies into the brain.79
A crucial question is whether, in the brain, inflammation is a component of the etiology of epilepsy, a consequence of seizures and cell damage, or both, and if inflammation can contribute to the progression of the disease.76
In the attempt to address these questions, preclinical investigations in experimental models of seizures focused first on the time-course of specific proinflammatory events occurring in the rodent brain after the induction of seizures.101 Much effort has been devoted to describe the regional distribution and cell-specific expression of proinflammatory molecules and their signaling in brain, vis-à-vis the patterns of seizure spread and the associated neuronal cell loss. Second, functional and pharmacologic studies have shown that cytokines can decrease the threshold for seizure induction and/or prolong the duration of seizures. Moreover, cytokines and downstream inflammatory mediators affect neuronal survival to injury, induce glial cell proliferation, modify BBB permeability to blood-borne molecules and cells, and inhibit neurogenesis.2 Pharmacologic evidence demonstrates that some anti-inflammatory treatments reduce seizures in experimental models and, in some instances, in clinical cases of epilepsy. All these findings support the hypothesis that inflammation in the brain plays a role in ictogenesis and, in some instances, also in neuronal cell loss, while also predicting its contribution to epileptogenesis. Direct proof for the latter is still lacking.
Several experimental findings have highlighted a dichotomous role for immune/inflammatory events in the CNS. Thus, these reactions can be also neuroprotective, and they thus constitute an adaptive, beneficial endogenous response to injury, similar to the classical immune/inflammatory responses to infection.2,63,92 In general, the deleterious effects of cytokines or other inflammatory mediators on neuronal excitability and cell survival appear to be mediated by their ability to provoke an extracellular rise of glutamate by actions on mechanisms of neurotransmitter release and/or reuptake, to potentiate the function of ionotropic glutamate receptors, and to enhance the production of mediators of oxidative stress (i.e., arachidonic acid and nitric oxide).2 However, cytokines can also induce the synthesis of nerve growth factor, ciliary neurotrophic factor, and insulin-like growth factor from astrocytes, all involved in brain repair.2 Other potential mechanisms of neuroprotection induced by cytokines include stimulation of antioxidant pathways and enhanced expression of manganese superoxide dismutase or calbindin, leading to an attenuated elevation of intracellular calcium (Ca2+) induced by tissue injuries. In summary, the final outcome of inflammation on brain function is highly dependent on the extent to which inflammatory mediators are produced, the length of time the tissue is exposed to inflammation, and the balance between the neurotrophic and inflammatory factors produced by the competent cells. It is also possible that the peripheral versus central origin of the cells and molecules involved in inflammation may play a role.
In this chapter, we review experimental and clinical evidence supporting the hypothesis that inflammation in brain may be a common thread playing a pro-epileptogenic role in various forms of epilepsy of different etiologies. Although the initial trigger of an inflammatory response in the brain remains still speculative (when the original description of the disease does not include the presence of specific pathogens), a large variety of insults that induce inflammation in the brain often result in the occurrence of seizures, and eventually in the onset of epilepsy, suggesting that an injury, even if subtle, occurring at birth or during lifetime may initiate a cascade of chronic inflammatory events in the CNS that contributes to the basis for the late onset of epilepsy (Fig. 1).
Understanding and awareness of the role of inflammation in epilepsy will increase our knowledge of the mechanisms underlying the etiopathogenesis of seizures and might open new perspectives for their pharmacologic treatment.
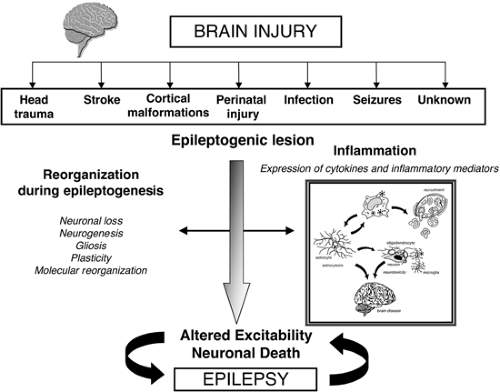 FIGURE 1. Schematic representation of the cascade of events that may be triggered by brain injury, eventually resulting in epilepsy. A large variety of injuries, even if subtle, occurring at birth or during a lifetime, may initiate anatomic, molecular, and synaptic reorganizations as well as a cascade of chronic inflammatory events in the brain that contribute to the late onset of epilepsy. Both innate and adaptive immune responses may play a role in initiating and consolidating inflammation in brain (exemplified in the boxed cartoon). The BBB is crucially involved in mediating the recruitment of cell components of the adaptive immune system, and alterations of the barrier properties may predispose the brain to the occurrence of seizures (see text for details). |
Experimental Models of Seizures
Expression Studies
During systemic infections (mimicked in rodents through the administration of lipopolysaccharide [LPS], a component of the gram-negative bacterial wall), the brain triggers an inflammatory response mounted to protect the host against infectious microorganisms. This phenomenon consists of an early inflammatory response (innate immunity) that can eventually progress to an adaptive immune response mediated by activated lymphocytes recruited from the blood.63,81 The CNS shows a well-organized innate immune reaction in response not only to infection but also to a variety of brain injuries. In particular, cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6, although expressed at very low levels in healthy brain tissue, are rapidly induced there following
ischemic, traumatic, and excitotoxic damage and seizures (for review see Allan2 and Vezzani101). Status epilepticus (SE), or recurrent seizures induced in rodents by electrical stimulation or application of chemoconvulsants, induces a rapid increase of proinflammatory cytokines and various markers of the innate immunity (e.g., nuclear factor [NF]-κB system, prostaglandins and their pathway enzymes, Toll-like receptors, chemokines, complement system) in glia and in neurons19,60,66,97 (Fig. 2). Increased production of inflammatory molecules in the brain has also been reported in genetic models of audiogenic seizures and in kindling.26,77 Markers of inflammation increase specifically in those brain regions involved in seizure onset and spread.
ischemic, traumatic, and excitotoxic damage and seizures (for review see Allan2 and Vezzani101). Status epilepticus (SE), or recurrent seizures induced in rodents by electrical stimulation or application of chemoconvulsants, induces a rapid increase of proinflammatory cytokines and various markers of the innate immunity (e.g., nuclear factor [NF]-κB system, prostaglandins and their pathway enzymes, Toll-like receptors, chemokines, complement system) in glia and in neurons19,60,66,97 (Fig. 2). Increased production of inflammatory molecules in the brain has also been reported in genetic models of audiogenic seizures and in kindling.26,77 Markers of inflammation increase specifically in those brain regions involved in seizure onset and spread.
The inflammatory response observed in mouse brain following pilocarpine-induced seizures differs from that described after systemic injection of LPS63,81,97,101: Thus, inflammatory molecules are first and predominantly expressed after endotoxemia in circumventricular organs, the choroid plexus, the leptomeninges, and along brain microvessels whereas microglia are involved to a minor extent and with a delay of several hours. Moreover, neurons do not typically express inflammatory markers after endotoxemia. Seizures induce a massive inflammatory response in parenchymal microglia and neurons (NF-κB and cyclooxygenase [COX]-2 are significantly expressed by neurons). Moreover, changes induced by endotoxemia are relatively short-lasting when compared to those observed after seizures. These observations suggest that inflammation induced by seizures in the brain results from complex neurophysiologic events that differ in their duration and in the cell populations involved from classical immune reactions triggered by infection.
The long-lasting stimulation of the innate immune response and related inflammatory reactions observed after seizures may eventually promote infiltration of lymphocytes and the establishment of acquired immunity in the CNS. Whether this phenomenon occurs is still controversial in animal models: There is indication of a late penetration of CD45+ monocytes into the brain parenchyma after seizures; however, Turrin and Rivest97 recently reported that markers of adaptive immunity, such as the production of IL-12 and interferon (IFN)-γ by activated T cells, are undetectable in the brain of pilocarpine-treated mice at least up to 72 hours after seizure induction. Accordingly, immunostaining for T cells, B cells, and natural killer (NK)-cells was negative in the brain of kainic acid–treated rats, although granulocytes and macrophages/monocytes were detected.21
The Role of Inflammation in Seizures
To address the functional consequences induced by inflammatory reactions on seizures and neuronal cell death, two main experimental approaches have been taken: (a) the intracerebral or systemic application of pro- and anti-inflammatory stimuli, and (b) the use of transgenic mice overexpressing proinflammatory cytokines in glia.
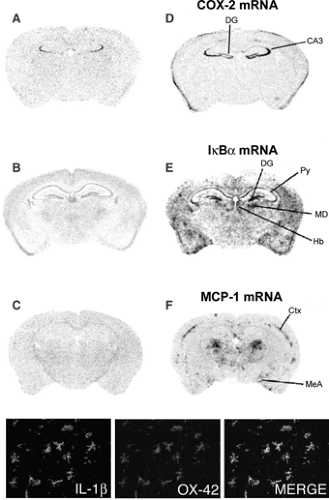 FIGURE 2. Seizure-induced expression of genes encoding proinflammatory molecules in rodent forebrain areas after seizures. Panels D through F show the increased hybridization signal of COX-2, IkBα, (an index of NFκB activation), and the chemokine monocyte-chemoattractant protein (MIP)-1, 24 hours after seizures induced by pilocarpine in mouse forebrain sections (adapted from Turrin NP, Rivest S. Innate immune reaction in response to seizures: Implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16:321–334, with permission). Very low or nondetectable expression was found in basal conditions (saline-injected mice), as depicted in corresponding panels A through C. The phenotype of the expressing cells was identified using selective markers: The majority of cells expressing these inflammatory mediators were infiltrating monocytes and parenchymal microglia; in some instances also endothelial cells were positive. Neurons also overexpressed COX-2 and IκB mRNA. Micrographs in bottom row depict IL-1β and OX-42+ microglia and their merged image, respectively. Ctx, cortex; CA3, pyramidal layer regions; DG, dentate gyrus; Hb, habenular nucleus; MD, mediodorsal thalamic nucleus; MeA, medial amygdala; Py, pyramidal cell layer. (See the color insert.) |
IL-1β significantly exacerbates seizure activity when intra-cerebrally injected in rodents shortly before the induction of
hippocampal seizures or SE.19,100,102 On the contrary, IL-1RA, a naturally occurring molecule that antagonizes the effect of endogenous IL-1β, has powerful anticonvulsant activity,103 and IL-1RA–overexpressing mice display a reduced susceptibility to seizures.102 These findings strongly suggest that an increase in brain levels of IL-1β has proconvulsant effects.
hippocampal seizures or SE.19,100,102 On the contrary, IL-1RA, a naturally occurring molecule that antagonizes the effect of endogenous IL-1β, has powerful anticonvulsant activity,103 and IL-1RA–overexpressing mice display a reduced susceptibility to seizures.102 These findings strongly suggest that an increase in brain levels of IL-1β has proconvulsant effects.
Recent data highlight the possibility that IL-1β signaling contributes critically to the hyperexcitability underlying febrile seizures (FS). Thus, intracerebroventricular injection of IL-1β reduces seizure threshold in 14-day-old mice subjected to hyperthermia22 or in immature rats exposed to LPS-induced fever31; IL-1β receptor–deficient mice or IL-1RA–injected rats were resistant to the induction of this kind of seizures.
It is important to note that changes in seizure threshold or duration induced by components of the IL-1β system are independent of alterations in core temperature.31 Hyperthermia-induced seizures can cause long-term effects on brain excitability in the absence of irreversible neuronal cell loss,9 therefore highlighting the possibility that the IL-1β system may be critically involved in fever-induced epileptogenesis.
Biochemical studies have shown that IL-1β can increase glutamatergic neurotransmission, and this action may mediate its effects on seizures: IL-1β enhances the calcium influx induced by N-methyl-D-aspartate receptor stimulation in pyramidal hippocampal cells by inducing the phosphorylation of the NR2B subunit of this receptor complex106; IL-1β inhibits glutamate reuptake by astrocytes111 and decreases the peak magnitude of γ-aminobutyric acid (GABA)-mediated currents in cultured hippocampal neurons.108
The effect of TNF-α on seizures depends on its brain levels and the receptor subtypes primarily involved: Relatively low doses of TNF-α reduce seizure activity by interacting with TNF-α p75 receptors,8 whereas mice overexpressing high amounts of TNF-α in glia show age-dependent spontaneous seizures and degenerative changes possibly mediated by p55 receptors.1 A direct interaction between TNF-α and α-amino-3-hydroxy 5-methylisoxazole 4-propionate (AMPA) receptors has been recently demonstrated in hippocampal neurons: This cytokine acting on p55 receptors regulates the cellular trafficking of AMPA receptors by inducing their membrane expression in a molecular conformation that amplifies the glutamate responses but reduces GABAA membrane receptors.91
Transgenic mice overexpressing IL-6 in glia show a profound increase in their sensitivity to glutamate-induced seizures, and neuronal loss of GABA- and parvalbumin-positive neurons was constitutively found in the hippocampi of these animals.84 Findings in transgenic mice suggest that a preexisting chronic inflammatory condition in the brain can predispose to the occurrence of seizures and promote neurologic dysfunctions. Accordingly, systemic administration of LPS to adult mice decreases their threshold to seizure induction, and this effect was blocked by anti-inflammatory drugs.86 IL-1β and TNF-α also play an important role in the sensitization of CNS to infection-related seizures in rodents caused by Shigella dysenteriae or Streptococcus pneumoniae.59,112
Table 1 Inflammatory markers and anti-inflammatory treatments in experimental models of seizures | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Limited information exists on the role in seizures for other cytokines such as fibroblast growth factor (FGF), IL-2, IL-3, or interferon (see Vezzani101 for details). Finally, sequential intrahippocampal infusion in rats of individual proteins of the membrane attack pathway of the complement system induced both seizures and neurotoxicity,110 implying that complement activation in the brain may contribute to seizures and cell death in diseases such as Rasmussen encephalitis.
Anticonvulsant Effects of Anti-inflammatory Strategies
Table 1 summarizes the data available on the effects of anti-inflammatory strategies on seizures. Nonsteroidal anti-inflammatory drugs (NSAIDs) can attenuate seizures; in particular, ibuprofen and indomethacin reduced penicillin-induced electrocorticographic and motor seizures in rats107; a similar effect was observed with paracetamol. Aspirin also protected mice from maximal electroshock (MES)- and pentylenetetrazol-induced seizures and potentiated the anticonvulsant action of diazepam and sodium valproate.90 In general, conflicting data are available on the effect of COX-1
and -2 inhibitors on seizures since prostaglandins can either reduce or promote seizures and affect neuronal cell survival differently depending on their specific type and receptor subtype interactions.6,14,101
and -2 inhibitors on seizures since prostaglandins can either reduce or promote seizures and affect neuronal cell survival differently depending on their specific type and receptor subtype interactions.6,14,101
Glucocorticoids (GCs) are potent inhibitors of the transcription of genes encoding most of the proinflammatory molecules, thus representing a critical endogenous negative feedback system with anti-inflammatory properties. Accordingly, a GC receptor inhibitor increased the inflammatory reactions induced by LPS in brain and enabled IL-1β and TNF-α to unveil neurotoxic effects. However, prolonged elevation of GCs (corticosterone) in the high physiologic range may induce a catabolically vulnerable state in neurons and result in an exacerbation of excitotoxic damage.85 This dichotomy may explain also the paradoxical proconvulsant effects of corticosterone and dexamethasone on seizures.51
Interestingly, valproate inhibits the LPS-induced activation of NF-κB and the subsequent production of TNF-α and IL-6 in monocytes and glioma cells. Carbamazepine was shown to decrease the LPS-induced production of prostaglandins and the activation of phospholipase A in rat glial cells.39,58 This evidence suggests that part of their anticonvulsant effects may be mediated by nonconventional anti-inflammatory actions.
Inflammation and Neuronal Cell Survival
When administered individually, cytokines do not directly lead to cell death but they can have synergistic effects (e.g., IL-1 plus TNF-α) that result in neurotoxicity involving both necrotic and apoptotic mechanisms. IL-1 exacerbates excitotoxic injury in vivo, whereas IL-1RA reduces excitotoxic, traumatic, and ischemic brain damage.2 Moreover, the extent of damage in the hippocampus after an excitotoxic insult appears to correlate significantly with the antecedent inflammatory cell infiltration and microglial activation, suggesting that neuronal damage can be at least in part caused by the preceding inflammation.21
Another cytokine effect that links them to epilepsy-related events is their inhibitory action on neurogenesis. In particular, neurogenesis is reduced by inflammation induced by radiation injury or LPS, whereas it is restored by indomethacin or inhibition of microglia activation by minocycline.23,61
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


