science revisited
Recent studies suggest that the type 1 interferon pathway is very active in dermatomyositis, providing opportunities for developing diagnostic biomarkers and therapeutic approaches.
Polymyositis
The term “polymyositis” encompasses a wide range of different diseases. Symmetric proximal weakness and certain muscle biopsy pathological features define this group, with considerable variation in how experts make this diagnosis. The alternative diagnosis of nonspecific myositis is appropriate for many patients. As a result of the marked heterogeneity of this category, unified mechanistic understanding is lacking. Many patients with “refractory polymyositis” have instead inclusion body myositis.
Inclusion Body Myositis
The term refers to an inflammatory myopathy that develops in mid or later life with a distinctive pattern of weakness involving asymmetric wrist flexor, finger flexor (Figure 3.1), and quadriceps weakness, and distinctive pathological features (inflammatory cells surrounding myofibers and rimmed vacuoles).
Figure 3.1. Asymmetric finger flexor weakness in inclusion body myositis. Patient is attempting to make a fist with both hands.
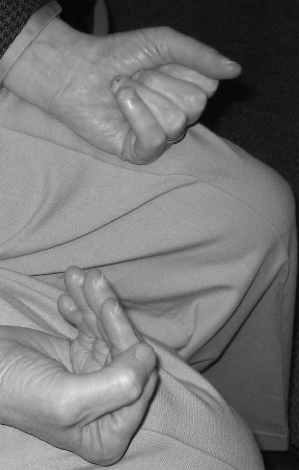
The mechanism of myofiber injury in IBM is poorly understood. Myonuclear abnormalities are present but have an unknown relationship with the mechanism of myofiber injury.
Clinical Evaluation
General
The diagnosis of inflammatory myopathy and the specific subtype is based on a combination of clinical presentation, laboratory studies, and pathological findings in muscle biopsy samples. In general, symptoms of acute or subacute (weeks to months) muscle weakness (difficulty arising from a low chair, climbing up or down stairs, getting into a car, washing hair, brushing teeth, or, in IBM, gripping objects) or skin rash (in dermatomyositis) are the presenting features. Patients presenting with prominent diffuse pain, often attributed to muscles, usually do not have an inflammatory myopathy. Pronounced lumbar lordosis and waddling gait, facial weakness, and scapular winging, signs of an indolent chronic myopathic process, should lead to considerations other than inflammatory myopathy.
Laboratory studies other than serum creatine kinase (CK) are of limited value to support or refute the diagnosis of inflammatory myopathy. The CK may be normal in active untreated DM. Serum “liver function tests” – aspartate aminotransferase (AST) and alanine aminotransferase (ALT) – may be elevated in inflammatory or other myopathies; these enzymes are present in muscle. Some patients with PM- or IBM-like clinical patterns of weakness may have associated HTLV-1 or HIV infection, so laboratory testing for these may be considered. Laboratory demonstration of autoantibodies, including antinuclear antibodies, anti-histidyl transfer RNA (anti-Jo-1) antibodies, anti-IFIH1 antibodies, and anti-Mi-2 antibodies may be helpful. The presence of anti-Jo-1 antibodies, associated with DM and PM, should raise suspicion for interstitial lung disease and prompt evaluation with pulmonary function tests, chest computed tomography (CT), and avoidance of methotrexate therapy because of the last’s potential pulmonary toxicity. Chest CT is also useful for consideration of sarcoidosis and as part of a malignancy evaluation for adults with DM.
Muscle biopsy and the pathological examination of the specimen obtained are important diagnostic procedures for patients with suspected inflammatory myopathies. In general, a mild-to-moderately weak muscle is optimal for biopsy. Good choices often are the biceps and vastus lateralis, but specific cases need to be considered individually.
Dermatomyositis
Dermatomyositis affects children and adults. Adult DM generally presents as subacute progressive painless proximal weakness, a skin rash, or both. Juvenile DM may present similarly or as an acute or subacute febrile illness followed by skin, muscle, or sometimes multisystem involvement.
The skin involvement in DM may have diverse manifestations, including: a heliotrope rash (purplish discoloration) on the eyelids; an erythematous rash on the face, neck, and anterior chest (“V-sign”), upper back (“shawl sign”), elbows, or knees; a purplish scaly papular rash on the extensor surface of the hands (Gottron’s papules); thickened and cracked skin on the dorsal and ventral surfaces of the hands (“mechanic’s hands”); and other changes. Subcutaneous calcinosis is a significant problem in juvenile DM and uncommon in adult DM. Cutaneous symptoms in DM have a high impact on lowering quality of life in patients and include prominent pruritus.
The pattern of proximal limb weakness in DM is not distinctive and does not distinguish DM from many other myopathies. Significant muscle asymmetries or prominent distal (forearm or lower leg) weakness together with skin rash should prompt consideration of sarcoidosis, for which clinical involvement similar to DM has been recognized. Normal serum CK may be present in patients with progressive disease and does not exclude the diagnosis. When elevated serum CK is present in DM, reductions generally occur with treatment and elevation with relapse.
Additional evaluation of adult patients with DM should be performed because of its association with two other important clinical syndromes: interstitial lung disease and malignancy. Pulmonary function tests, chest CT, and laboratory testing for the presence of anti-histidyl-tRNA antibodies (anti-Jo-1 antibodies) should be considered in all patients with DM. Malignancy has been estimated to be associated with 6–45% of adult patients with DM, with age-associated increased risk particularly in women aged over 40. A malignancy evaluation, including physical examination (skin examination, breast and pelvic examinations in women, and testicular and prostate examination in men), blood studies (complete blood count, liver function tests, lactate dehydrogenase, prostate-specific antigen), stool for occult blood, CT (chest, abdomen, and pelvis), and colonoscopy should be considered in every adult patient with a new diagnosis of DM.
 tips and tricks
tips and tricks
- Malignancies are associated with 6–45% of patients with DM.
- A malignancy evaluation is essential in patients diagnosed with DM.
Muscle biopsy is an important diagnostic procedure in DM. The most supportive diagnostic features of muscle biopsies for DM evident in routine clinical studies are the presence of perifascicular atrophy and the absence of multiple myofibers surrounded by inflammatory cells. Perifascicular atrophy refers to the presence of small myofibers that are slightly darker and bluish in color in hematoxylin and eosin sections, typically located at the edges of fascicles (Plate 3.1).
Inclusion Body Myositis
Inclusion body myositis affects adults in middle and later life. Onset before age 50 occurs in 18–20% of patients and then mostly after age 50. Diagnosis has historically been frequently delayed by a mean of 5–8 years from symptom onset.
The clinical presentation of IBM is distinct from that of other inflammatory myopathies. Atrophy and weakness of wrist and finger flexors and quadriceps are distinctive, and physical examination should focus on careful testing of these muscle groups. Comparison of wrist and finger extensors with corresponding flexors may demonstrate the greater involvement of the flexors and asymmetries (see Figure 3.1). Relative preservation of deltoids, in comparison to the forearm flexors, can be impressive, in marked contrast to the pattern of weakness seen in DM and PM. Involvement of tibialis anterior may also be distinctive in IBM. Dysphagia can be a significant problem with a prevalence estimated as high as 66%.
Serum CK is only modestly elevated; research criteria have proposed diagnostic criteria of an upper limit of 12 times the upper limit of normal, although patients with higher values, up to 16 times the upper limit of normal, have been reported.
On muscle biopsy, the presence of multiple myofibers surrounded by inflammatory cells and many myofibers with rimmed vacuoles is highly supportive of a pathological diagnosis of IBM. Both IBM and PM (see below) may have similar patterns with respect to the location of inflammatory cells as seen in routine studies. The pattern of inflammatory cells deep within fascicles surrounding and sometimes invading myofibers (Figure 3.2) is distinct from that of DM. What distinguishes IBM from PM in light microscopic examination is a sufficient number of rimmed vacuoles as well as the presence of signs of a chronic indolent process in IBM such as hypertrophied fibers and fibrosis. Difficulties with diagnosis occur in patients with typical clinical features but few inflammatory cells or with few rimmed vacuoles.
 tips and tricks
tips and tricks
- Unlike PM and DM, IBM presents with a distinct pattern of muscle weakness.
- Finger flexors, wrist flexors, and quadriceps are selectively involved in IBM and associated with a “scooped-out” atrophic forearm flexor compartment and atrophy of quadriceps.
- In addition to the characteristic limb weakness, dysphagia occurs in most IBM patients.



