Intramedullary Spinal Cord Tumors
Primary spinal cord tumors account for approximately 2 to 4% of all central nervous system neoplasms.1–3 The percentage approaches 35% in the pediatric population as well as in young adults,1 and roughly 100 to 150 cases of pediatric intramedullary spinal cord tumors (IMSCTs) are diagnosed in the United States each year.1 Ependymomas, astrocytomas, and gangliogliomas account for approximately 80% of IMSCTs.4–8
The development of modern microsurgical techniques, operative equipment, neuroimaging, and intraoperative neurophysiology has elevated aggressive resection of these lesions to the standard of care.1,5,6,9,10 As a result, these patients have experienced improvement in long-term survival and quality of life, although late recurrences do occur despite adequate surgical resection. Adjuvant therapy, including radiation and chemotherapy, has resulted in only modest improvement in survival. Tumor location, the patient’s age, pathology, and the ability to achieve a gross total resection (GTR) determine whether any adjuvant treatment is necessary.
46.1 Epidemiology
IMSCTs are found throughout the entire neuraxis. In the pediatric population, the majority of these lesions occur in the cervical and cervicothoracic region,7,8,11,12 whereas in adults the incidence of intradural extramedullary tumors is increased, and thus the number of lumbar lesions that are diagnosed.5 Astrocytomas are the most common IMSCTs found in children5,6 but can occur at any age.2,7,13 In patients younger than 10 years of age, more than 90% of spinal cord tumors are glial in origin, although this percentage declines to 60% of patients by the adolescent years,5,12 with an increased prevalence of ependymomas.
Ependymomas are believed to arise from the ependymal lining of the ventricles and central canal in the brain and spinal cord. There is an association with neurofibromatosis, but most cases are spontaneous. In children, ependymomas are the second most common lesion following astrocytomas and typically occur in the cervical region.5,13–15 In children, more than 90% of the tumors are intracranial in origin, and in one series no spinal cord ependymomas were diagnosed in children younger than 3 years of age.16
46.1.1 Associations with Neurofibromatosis
Neurofibromatosis type 1 (NF-1) and neurofibromatosis type 2 (NF-2) are both associated with tumors of the central nervous system.17 NF-1 is an autosomal-dominant disorder with complete penetrance. It has a prevalence of 1 per 3,000. NF-2 is a rarer disease, with a prevalence of 1 per 40,000 people.18 It is caused by a mutation of the tumor suppressor gene on chromosome 22 called merlin or schwannomin.5
Dow and colleagues showed that patients with NF-1 were more likely to present with IMSCTs that were astrocytomas, whereas patients with NF-2 more commonly had IMSCTs that were ependymomas.19 In the series of Lee and colleagues, the incidence of IMSCT in the total population of patients with neurofibromatosis was 19%.18 In the series of Malis et al, among 41 patients with NF-2 and 99 spinal tumors, 14 presented with intramedullary tumors.20 NF-2 is more commonly associated with schwannomas and meningiomas, but there is also an association with IMSCTs. Although patients with NF-2 comprise only 0.03% of the population, they are overrepresented among patients who have IMSCTs (2.5%).18 Additionally, Birch and colleagues showed that 71% of patients with intramedullary ependymomas who did not meet the clinical criteria for NF had mutations in the NF2 gene21 (▶ Table 46.1).
| Neurofibromatosis type 1 | Neurofibromatosis type 2 | |
| Genetic mutations | von Recklinghausen | merlin, schwannomin |
| Chromosome 17 | Chromosome 22 | |
| Associated intramedullary spinal cord tumors | Astrocytoma | Ependymoma |
| Other associated central nervous system tumors | Nerve root neurofibromas, optic nerve gliomas, meningiomas | Bilateral acoustic neuromas and schwannomas |
46.2 Presentation
As in patients with other neuraxis lesions, symptoms depend on the location of the tumor. Most commonly, patients present with pain, followed by gait deterioration, weakness, bowel and/or bladder dysfunction, and sensory disturbance.6,10,15 Sensory changes also present differently across the age groups. In young children, localization can be difficult. Their symptoms may manifest only through general irritability or an avoidance of certain positions. Older children sometimes describe a dull, aching, nonspecific type of nocturnal pain. Adolescents may attribute their pain to normal growth and may not present until they demonstrate weakness or an atypical focal finding, such as a subtle change in gait or frequent falling. The pain is typically worse in the supine position as venous congestion distends the dura, causing nocturnal pain. Adults, conversely, will recognize a deterioration of normal function and present with a more classic sensory disturbance.15,22
Children also manifest other typical findings differently from adults. Weakness may be difficult to detect in young children, who are innately clumsy as they are learning to walk. Bowel and bladder dysfunction may also be difficult to detect in infants who are not toilet-trained. However, in a child who has been previously toilet-trained, loss of bowel and bladder control can be a manifestation of an underlying pathology.6,10
Ependymomas, arising from the ependymal cells of the central canal, are well-circumscribed, slow-growing tumors that are centrally located and cause symmetric expansion of the spinal cord.5,23,24 Patients typically present with bilateral dysesthesias at the level of the tumor, as well as paresthesias, radicular pain, bowel and bladder dysfunction, and other sensory disturbances.15,22,24 Children commonly present with pain, weakness, gait abnormality, torticollis, or progressive kyphoscoliosis.1,6 Hydrocephalus is more commonly found in pediatric patients with spinal cord ependymomas or myxopapillary ependymomas than in those with other IMSCTs and requires shunting occasionally. However, hydrocephalus develops in fewer than 10% of patients, and the etiology is an increased protein content in the cerebrospinal fluid (CSF), arachnoidal fibrosis, and subarachnoid metastasis.10,25 Acute decline can follow an intratumoral hemorrhage.24
Patients with IMSCT astrocytomas have symptoms similar to those of patients with ependymomas. Worsening of motor symptoms tends to be earlier in the disease process in these patients than in those with ependymomas. Spinal deformity can be common and presents in up to 30% of patients.2,26 High cervical lesions with involvement of the medulla can present with bulbar symptoms, including vomiting, choking, dysphagia, frequent respiratory infections secondary to chronic aspiration, dysarthria, dysphonic speech, and sleep apnea. In younger children, these deficits can manifest as failure to thrive.
46.3 Imaging Characteristics
Magnetic resonance (MR) imaging has dramatically changed the way IMSCTs are evaluated because this is typically the only imaging modality needed to establish the diagnosis. Intramedullary lesions appear as an area of cord expansion on MR imaging and are often associated with cysts or syringomyelia. Tumors may appear isodense on T1-weighted images, and tumoral cysts are hyperintense on T2-weighted images because of the high protein content. The majority of tumors will enhance after gadolinium administration, and differences in enhancement may suggest certain histologies.27 Although a precise histologic diagnosis is not possible via imaging, the lesions do tend to follow typical patterns on imaging27–29 (▶ Table 46.2).
| Astrocytomas | Ependymomas | |
| Location | Eccentric location Cervicothoracic spine Can extend holocord | Central location Cervical spine |
| T1 | Iso- to hypointense | Iso- to hypointense |
| T1 with contrast | Heterogeneous contrast enhancement with ill-defined borders | Symmetric contrast enhancement with sharply defined borders |
| T2 | Hyperintense | Hyperintense |
| Cysts | Less common than with ependymomas except for juvenile pilocytic astrocytomas | Three types of cysts: tumoral cysts from necrosis and hemorrhage, syrinx formation, rostral and caudal cysts from reactive tumor products |
| Unique findings | Peritumoral edema, more commonly associated with syrinx formation | “Cap” sign, in which areas of low signal density appear on either tumor border that are due to hemosiderin deposits from secondary, chronic hemorrhage |
46.4 Differential Diagnosis
Overall, the most common lesions are primary glial tumors. Astrocytomas are the most commonly occurring lesion, followed closely by ependymomas, gangliogliomas, oligodendrogliomas, neurodevelopmental tumors, and malignant gliomas.5 Other, nonneoplastic lesions may also occur, such as cavernous malformations and arteriovenous malformations, as well as infectious and inflammatory pathologies.
46.4.1 Astrocytomas
IMSCT astrocytomas arise from transformed astrocytes that then infiltrate the spinal cord. They are histologically identical to intracranial astrocytomas and are graded on the same World Health Organization (WHO) scale: pilocytic lesions (grade I), low-grade astrocytomas (grade II), anaplastic astrocytomas (grade III), and glioblastoma multiforme (grade IV).
Astrocytomas are the most common IMSCTs occurring in children and account for nearly 40 to 60% of lesions.1,6,10,15,22 They are the second most common type in adults, with a frequency of 20 to 30%2,5 (▶ Fig. 46.1). Compared with their intracranial counterparts, they are more benign in presentation and are more commonly low-grade lesions at diagnosis. High-grade astrocytomas comprise 10 to 15% of pediatric spinal cord tumors.2,5 They more commonly occur in the cervical and thoracic region in children (▶ Fig. 46.2). Juvenile pilocytic astrocytomas may be associated with large tumoral cysts; the solid component is often well localized and should be the area that is targeted surgically.9,22
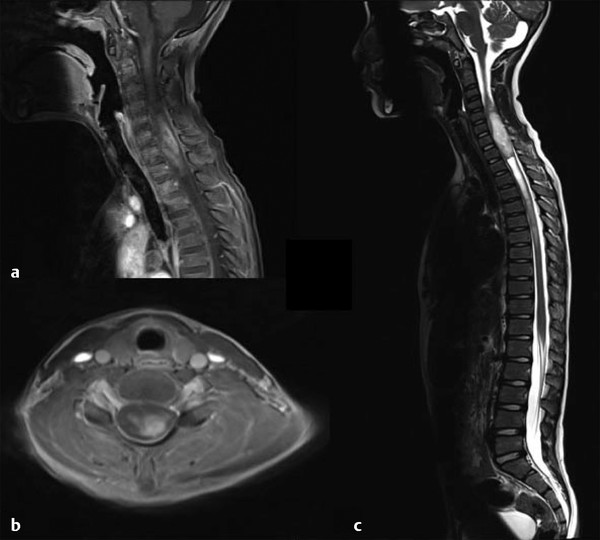
Fig. 46.1 Magnetic resonance imaging in a patient presenting with progressive myelopathy over many months. The histologic diagnosis was World Health Organization grade I astrocytoma. (a) Sagittal T1-weighted image demonstrating heterogeneous enhancement with expansion of the spinal cord. (b) Axial T1-weighted image showing contrast enhancement eccentrically located within the spinal cord. (c) Sagittal T2-weighted axial image showing diffuse hyperintensity with caudal syrinx formation.
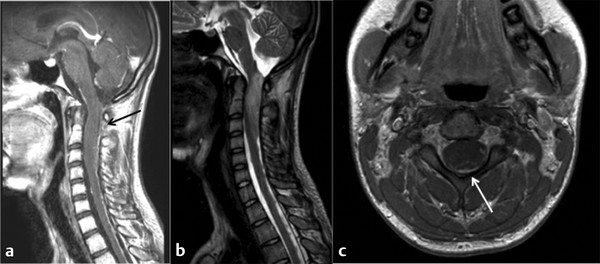
Fig. 46.2 Magnetic resonance imaging in a patient presenting with the acute onset of difficulty walking. The histologic diagnosis was World Health Organization grade IV astrocytoma. (a) Sagittal T1-weighted image shows minimal enhancement of the tumor itself, with the black arrow indicating diffuse pial enhancement. (b) Sagittal T2-weighted image showing hyperintensity and edema throughout the cervicomedullary junction. (c) Axial T1-weighted image, with the white arrow showing contrast enhancement along the pial border.
Although genetic alterations are well described for intracranial tumors, very few data exist for spinal cord lesions. However, we assume that there is a correlation because some IMSCT astrocytomas are clustered with inherited syndromes: Li-Fraumeni syndrome, Turcot syndrome, tuberous sclerosis, Maffucci-Ollier disease, and NF.5 Although less common than in adult tumors, chromosomal changes in 7, 22, and 10 and molecular alterations of EGFR, PTEN, and IDH1 have all been identified in a subset of pediatric gliomas and may contribute to the transition from low-grade to high-grade astrocytoma.12,30 However, the genomic changes in most pediatric astrocytomas differ from those in adults, suggesting that these tumors may arise from different molecular pathways, which is an area of intense investigation.
46.4.2 Ependymomas
Ependymomas account for 4 to 6% of primary central nervous system tumors, with approximately 30% of these lesions being intraspinal.5,12 They can be divided into four subtypes via the WHO classification: subependymoma, myxopapillary ependymoma (grade I), benign or “classic” ependymoma (grade II), and anaplastic ependymoma (grade III). Subependymomas rarely occur in the spinal cord and are mostly intraventricular. Myxopapillary lesions arise from the filum terminale or conus medullaris and are thus found in the lumbar region 20 to 25% of the time, most commonly in adults.12 Most benign and anaplastic lesions occur in the cervical region. They tend to present in adults between the ages of 30 and 50 years but are the second most common lesion in the pediatric population (▶ Fig. 46.3). These tumors are relatively avascular but typically receive their blood supply from branches of the anterior spinal artery, and care should be taken to preserve the normal vasculature during removal.
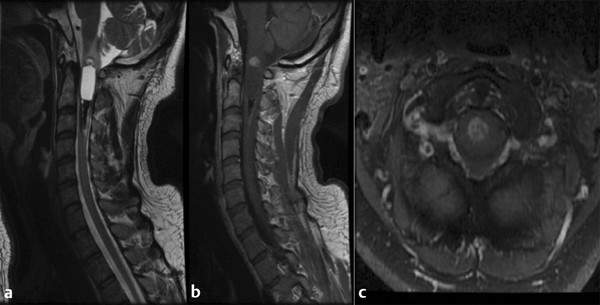
Fig. 46.3 Magnetic resonance imaging in a patient presenting with neck pain over a period of several years who was originally followed conservatively and showed progression. The histologic diagnosis was ependymoma. (a) Sagittal T2-weighted image showing hyperintensity in a central location. Both rostral and caudal cysts show evidence of prior hemorrhage and the classic “cap” sign. (b) Sagittal T1-weighted image shows minimal enhancement of the tumor. (c) Axial T1-weighted image showing central heterogeneous enhancement.
Approximately 75% of all ependymomas have some chromosomal rearrangement or change.5 Mutations of the genes on chromosome 22 associated with NF-2 (merlin) are the most common and occur about 40% of the time. Other chromosomes that have been implicated are chromosome 6 (30.3% of the time), chromosome 9 (27.3% of the time), and chromosome 1712,18,19. The molecular and genetic events that distinguish spinal ependymomas from intracranial ependymomas are more defined than those for spinal and intracranial astrocytomas. Methylation patterns in certain genes, including the tumor suppressor gene HIC1, have been implicated in intracranial lesions compared with spinal ependymomas.5,12,30 Most recently, Johnson and colleagues have shown distinct genetic signatures for intracranial ependymomas compared with ependymomas originating in the spinal cord. Members of the Notch and Sonic Hedgehog pathways are overexpressed in intracranial ependymomas, whereas overexpression of the Homeobox-containing family of genes (HOX) is implicated in spinal cord ependymomas.31 These authors have also found that ependymomas are derived from regionally specific stem cells bearing a radial glial cell phenotype.31
46.4.3 Gangliogliomas
Gangliogliomas are composed of both neuronal and glial cells. The glial elements are typically astrocytes, and the neoplastic neurons are large and mature in appearance. The frequency of these tumors among IMSCTs has been reported to be as high as 27% in some series,11,32 and lesions can be infiltrative (▶ Fig. 46.4). They are slow-growing and occur in the cervical and upper thoracic spine in an eccentric location. Given their slow growth, they can be associated with bony erosion or scalloping. On imaging, they have areas of mixed signal on T1-weighted images, with heterogeneous enhancement that extends to the surface of the spinal cord, and are associated with prominent tumoral cysts.11,32
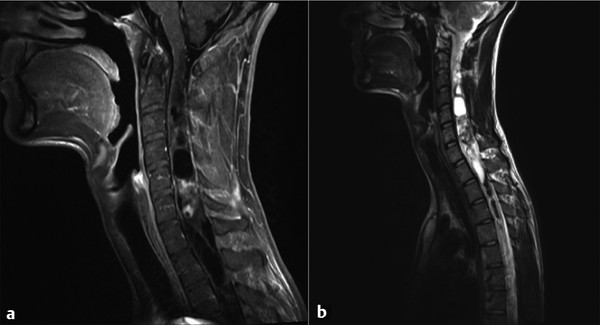
Fig. 46.4 Magnetic resonance imaging in a patient presenting with myelopathy over several years. The histologic diagnosis was ganglioglioma. (a) Sagittal T1-weighted image demonstrating heterogeneous nodular enhancement. (b) Sagittal T2-weighted image demonstrating extensive cystic components both rostral and caudal to the nodular area.
46.4.4 Lipomas
Lipomas account for 1% of IMSCTs and arise from embryonal clefts. Although they are commonly thought to be associated with spinal dysraphism, they are indolent in nature, and the presentation is often delayed until adulthood. They are commonly located in the thoracic spinal cord12 and are histologically identical to normal adipose tissue. They may grow, like any other fatty deposit in the body, and patients may manifest myelopathic signs following spurts of growth or weight gain. Lipomas typically appear as hyperintense lesions on T1-weighted imaging without contrast. Although these lesions are distinct from the spinal cord, they are densely adherent, so that total removal without neurologic deficits is difficult.27
46.4.5 Hemangioblastomas
Hemangioblastomas are relatively rare in the pediatric population, but if found, they are typically associated with von Hippel-Lindau disease.5,33 They are located on the dorsal surface of the spinal cord and are associated with large feeding and draining vessels.
46.4.6 Others
Lymphomas and metastatic lesions are more common in the adult population. Lymphomas are rare and are associated with AIDS. They are almost uniformly found in adults. Vascular malformations, including cavernous angiomas, are similar to their intracranial counterparts. They typically present in young adulthood, either with multiple small hemorrhages leading to myelopathy or with an acute hemorrhage that leads to an acute neurologic decline.12 They account for 1 to 3% of all IMSCTs. Metastatic lesions are also almost always found in adults and can occur from both hematogenous spread and direct leptomeningeal invasion.12 Other IMSCTs include dermoids, teratomas, oligodendrogliomas, and nonneoplastic lesions, such as sarcoid and multiple sclerosis.
Other intramedullary spinal lesions include inclusion tumors and cysts, nerve sheath tumors, neurocytomas, and melanocytomas. Approximately 4% of IMSCT are found to be non-neoplastic lesions.5,12
46.5 Treatment Strategy
Surgical intervention is the primary treatment of choice for IMSCTs; the goal is complete resection because progression-free survival (PFS) is good when this occurs.8,11,13,14,24 Despite multiple trials and retrospective reports, little progress has been made with adjuvant therapy to improve overall survival (OS). Radiation treatment is more widely accepted for patients with incomplete resection and for those with astrocytomas. The clinical benefits of GTR are best established for ependymomas.
The natural history of many IMSCTs is more benign than that of their intracranial counterparts. These patients are often neurologically stable for years with simple observation. However, most surgeons agree that open surgery is indicated because the lesions undoubtedly eventually progress to neurologic deficit.5,10,12,22 The outcome for low-grade spinal cord astrocytomas is better in children than in adults, although the prognosis for astrocytoma is not as favorable as that for ependymoma.8,10,23,34 Several series have shown that patients with good preoperative function have a better recovery following operation than those with more severe preoperative symptoms.7,8,15 One hypothesis is that the spinal cord tissue is more plastic in children than in adults, and this portends a better prognosis.
46.5.1 Surgery
The goal of surgery for low-grade IMSCTs remains GTR with preservation of neurologic function.3,8,9 Although the data are more controversial for astrocytomas, several series with data for grade II and grade III lesions have demonstrated an improvement in OS and PFS with aggressive resection of the tumor.3,8,22 There are no class I studies for the treatment of IMSCTs.
Maintaining the patient’s neurologic function remains an important goal of surgery. Patients with minimal preoperative symptoms have a greater potential for improvement postoperatively. If the tumor is infiltrative, then decompression of the adjacent spinal cord and the avoidance of neurologic dysfunction should be the goal.1,12,23 Closely adherent tumor should not be forcibly removed.6 Surgery should ideally occur before the patient has experienced a major neurologic decline because the potential to stabilize or to regain function is limited in these patients.
Astrocytomas are typically eccentrically located several millimeters beneath the dorsal surface of the spinal cord. Radical resection is more controversial because they are infiltrative in nature. However, an excellent prognosis has been reported for patients with low-grade astrocytomas,2,7,22 and a radical resection will improve PFS and OS for these patients. These tumors harbor microscopic disease beyond what is visible at the time of surgery, and thus the removal of all cellular atypia is unlikely. Therefore, resection should be continued until the interface between normal spinal cord and tumor tissue becomes difficult to differentiate, so as to minimize the resection of normal neurologic pathways.6,8 The tumors are typically resected from the inside out until the glia–tumor interface is identified by the change in color and consistency of adjacent spinal cord. Neuro evoked potentials are critical to guide the extent of resection in the event that motor pathways impede resection.
Like intracranial lesions, high-grade astrocytomas have a poor prognosis, which is not altered by surgical resection.1,7,22 Despite resection, they recur rapidly and metastasize throughout the subarachnoid space within a few months. An early biopsy should be taken during surgery if there is any concern for atypia because this will influence how aggressively the tumor will be removed. Evidence of a high-grade lesion, such as a glioblastoma, portends a poor prognosis despite surgical resection. In this case, if there is a cystic component that is causing mass effect, then decompression may lead to some temporary relief of symptoms. However, the patients do poorly and have a median survival of less than 2 years even with adjuvant therapy.
Ependymomas are more distinct from the surrounding spinal cord and have a clear cleavage plane that facilitates resection.14 They are central in location, and given their cystic nature, the cleavage plane can be further defined at the poles of the tumor. The goal at surgery is to minimize trauma and traction on the normal spinal cord.6,9,14 Because PFS correlates with the extent of resection, the surgeon should pursue a GTR.5,12,22 However, if the interface between tumor and normal spinal cord becomes indiscernible, then the resection should be halted because neurologic function should be preserved.
46.5.2 Intraoperative Management
Surgical technique is determined by the tumor size and histologic diagnosis at the time of biopsy. A laminoplasty can be performed to expose the extent of the solid component of tumor.7 Although laminoplasty has not been definitively shown to reduce the progression of deformity, it has been shown to lower the CSF leakage rate and to help preserve normal anatomical planes.35 If there is concern for the accuracy of the surgical exposure and identification of the tumor, intraoperative ultrasound may be obtained to optimize the size of the laminoplasty, dural opening, and myelotomy. Ultrasonography can also be an important adjunct for identifying the solid and cystic components of the tumor and for determining the depth of resection.
Intraoperative electrophysiologic monitoring is critical to establish the extent of resection during removal of an IMSCT. Somatosensory evoked potentials (SSEPs) measure the afferent conduction of impulses from a peripheral nerve to the brainstem or cerebral cortex. Degradation of the signal results from insult to the sensory pathways, such as the dorsal columns or anterolateral tracts.36,37 SSEPs are recorded as an average of many signals, and this can delay the detection of injury. Therefore, motor evoked potentials (MEPs) and epidural electrodes give a better real-time estimate of the integrity of motor pathways. MEPs record a peripheral response to cortical stimulation and are shown as an “all-or-none” interpretation profile. The epidural MEP, or D-wave, measures the direct activation of large-diameter corticospinal axons through stimulation of the spinal cord with an electric current. The amplitude of this signal correlates with the number of intact descending motor units or functioning axons.36–38 The epidural electrodes are placed both rostral and caudal to the tumor following the laminectomy.
The intraoperative monitoring of D-waves can make it possible to predict the postoperative functional outcome of patients.38 Both MEP and D-wave monitoring can predict spinal cord damage and transient postoperative effects. If the muscle MEPs are lost but the D-wave is preserved during surgery, then the patient typically has a transient deficit postoperatively. A decrement of 50% in the amplitude of MEPs is an indication to halt surgical resection because this is a measure of a temporary as well as a potentially permanent deficit8,36–38 (▶ Table 46.3). These techniques rely on computer averaging, so there can be a brief delay of 10 to 60 seconds between the time of injury and visualization of a declining response amplitude.
| D-wavea | Muscle motor evoked potentialb | Postoperative motor status |
| Decreased < 50% | Unchanged | Unchanged |
| Decreased < 50% | Unilateral or bilateral loss | Transient motor deficit |
| Decreased > 50% | Bilateral loss | Prolonged motor deficit |
| aD-wave measures the direct activation of large-diameter corticospinal axons through direct stimulation of the spinal cord by an epidural electrode. bMuscle motor evoked potentials measure the peripheral response to cortical stimulation as an “all-or-none” interpretation. | ||
This technology is not without limitations because intraoperative mapping can be influenced by other factors. Anesthetic agents can affect whether nerves can be mapped. Close discussion with the anesthesiologists should be conducted to minimize the intermittent administration of boluses of intravenous anesthetics and of halogenated anesthetics, and to avoid hypothermia. Paralytic agents should also be avoided because they can influence the ability to record MEPs. If the patient has significant weakness preoperatively, the signals can be difficult to obtain and follow.
46.5.3 Deformity
Spinal deformity occurs in conjunction with IMSCTs even if there are no neurologic signs or symptoms. Some children can even present with scoliosis, and up to 37% of children will require stabilization or a brace.14 This high rate of instability relates to the relative laxity of the ligaments, the horizontal orientation of facet joints, and the dynamic growth of the osseous spine.14,39 Given the low prevalence of instability, prophylactic instrumentation is not used. However, several risk factors are associated with progression to fusion, included age younger than 13 years, involvement of the thoracolumbar junction, tumor-associated syrinx, and preoperative scoliotic deformity1,6,7,26 (see box “▶ Predictors of Progressive Spinal Deformity Requiring Fusion after Resection of Intramedullary Spinal Cord Tumors”). The incidence of progressive kyphoscoliosis in the thoracic spine and swan neck deformities in the cervical spine is higher in children than in adults.7,26 The cervical region is more prone to destabilization than the thoracic or lumbar region. Deformity is also related to the extent of the bony decompression. Radiation and tumor-induced paraspinal muscular weakness further destabilize the body’s ability to compensate for bony loss.26,39
Predictors of Progressive Spinal Deformity Requiring Fusion after Resection of Intramedullary Spinal Cord Tumors
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


