Chapter 30 Introduction to Genetics
In the broadest sense, genes are simply units of hereditary information; the genome is the totality of all the hereditary information in a cell or organism; and genetics may be defined as the study of genes and genomes. With the advent of modern molecular biology and the Human Genome Project, all aspects of genetics have come to play a more prominent role in the day-to-day evaluation and management of children with neurologic diseases, most of which have a genetic basis. This chapter presents a brief synopsis of the most important principles of genetics, to serve as background for information presented elsewhere in this text. More detailed information on genetics is available in many excellent textbooks, such as Genetics in Medicine [Nussbaum et al., 2007], Genes IX [Lewin, 2007], and Human Molecular Genetics [Strachan and Read, 2010]. Other resources are available from the National Center for Biotechnology Information website (Table 30-1).
Table 30-1 Genetic Information Websites
| Site | Internet Address |
|---|---|
| NCBI1 Genetic Disease Websites | |
| GeneTests, GeneReviews2 | http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
| OMIM3 | http://www.ncbi.nlm.nih.gov/omim/ |
| NCBI1 Genome Data Websites | |
| NCBI1 homepage (Entrez) | http://www.ncbi.nlm.nih.gov/ |
| dbGaP Genotypes and Phenotypes | http://www.ncbi.nlm.nih.gov/gap |
| dbSNP (SNP database) | http://www.ncbi.nlm.nih.gov/snp/ |
| Other Genome Data Websites | |
| Ensembl Human Genome Browser | http://uswest.ensembl.org/index.html |
| HUGO4 | http://www.genenames.org/index.html |
| DOE5 Genomics Websites, includes Human Genome Project | http://genomics.energy.gov/ |
| UCSC Genome Bioinformatics6 | http://genome.ucsc.edu/ |
1 National Center for Biotechnology Information.
2 Disease summaries in GeneReviews are authored by experts and peer-reviewed, and so are typically highly accurate and up to date.
3 Disease summaries in OMIM (Online Mendelian Inheritance in Man) are done by staff with oversight and contain both dated and new data; all information from OMIM should be confirmed from a second source.
4 The HUGO Gene Nomenclature Committee website established accepted names for human genes.
5 U.S. Department of Energy Office of Science websites, which include the Human Genome Project website.
6 University of California–Santa Clara Genome Bioinformatics site, which contains the most widely used human genome browser, sometimes called “Golden Path.”
Molecular Basis of Heredity
Structure and Function of DNA
DNA is a large polymer or macromolecule composed of linear sequences of simple repeating units. The specific sequence of these units contains all of the genetic information of an individual cell or organism. The structure of DNA in its native state was deduced by Watson and Crick in 1953 [Watson and Crick, 1953]. The basic repeating unit of DNA is the nucleotide, which consists of a five-carbon sugar known as deoxyribose; a phosphate group; and a nitrogen-containing base, which may be either a purine or a pyrimidine (Figure 30-1A). In DNA, the purine base may be either adenine (A) or guanine (G), and the pyrimidine base may be either thymine (T) or cytosine (C). Nucleotides polymerize into long chains by formation of phosphodiester bonds between the 5′ carbon position of one deoxyribose molecule and the 3′ carbon of the preceding deoxyribose molecule (Figure 30-1B).
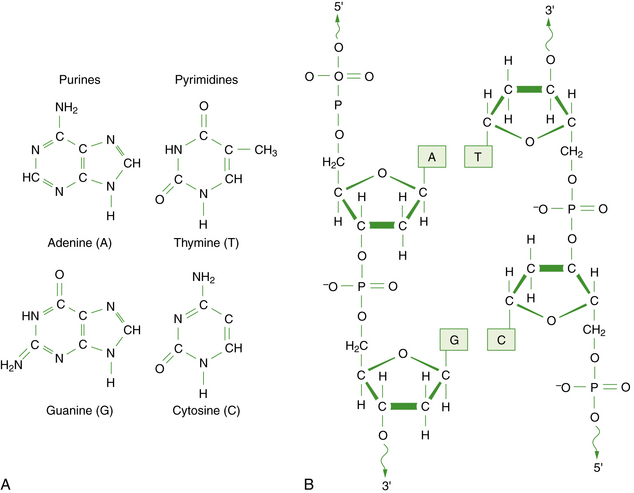
Fig. 30-1 The chemical structure of DNA.
A, The four bases of DNA. B, The sugar-phosphate backbone and 3′–5′ phosphodiester bonds.
Each DNA molecule consists of two strands of nucleotides that are held together by weak hydrogen bonds between pairs of bases: A pairs only with T, and G pairs only with C. These paired units are known as basepairs (bp). In the native state, the two strands wind around each other to form a double helix that resembles a right-hand spiral staircase, with two unequal grooves known as the major and minor grooves (Figure 30-2). A single turn of the helix measures 3.4 nm and contains ten nucleotides. Each strand has a directionality imparted by the deoxyribose sugar backbone. Adjacent nucleotides are linked by phosphodiester bonds between the 5′ and 3′ carbon atoms of the sugar residues, so that one end of the DNA strand has an unlinked 5′ carbon (the 5′ end) and the other end of the strand has an unlinked 3′ carbon atom (the 3′ end). The two strands are antiparallel – that is, they run in opposite directions so that the 5′ end of one strand is paired with the 3′ end of the other. Within living cells, DNA is associated with proteins and supercoiled into more complex structures known as chromosomes, which are described later in the chapter.
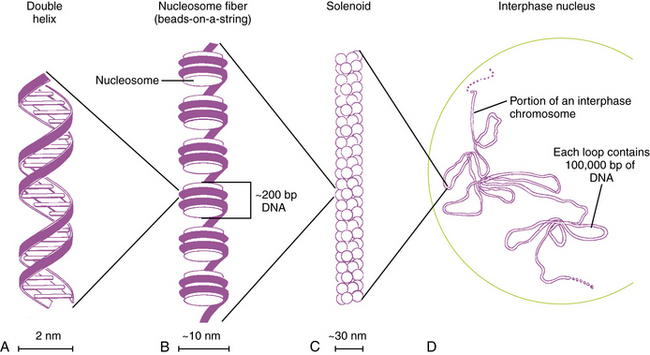
Fig. 30-2 Packaging of DNA by structural proteins.
(Modified from Thompson MR et al. Genetics in medicine, 5th edn. Philadelphia: WB Saunders, 1991.)
Five distinct DNA polymerases have been isolated in mammalian systems, including human cell cultures (Table 30-2). They are able to copy DNA only by adding nucleotides to the 3′ end of the growing chain, so DNA can elongate only in the 5′ to 3′ direction. Thus, the template DNA can be read only in the reverse, or 3′ to 5′, direction. As DNA is unwound, the replication fork necessarily unwinds one strand in the 3′ to 5′ direction and the other in the 5′ to 3′ direction. The 3′ to 5′ or leading strand is replicated in a continuous fashion at the replication fork by DNA polymerases α(I), which primes the reaction, and δ(III), which synthesizes the DNA chain. The new strand is complementary and so elongates in the opposite, or 5′ to 3′, direction.

Structure and Function of RNA
RNA differs chemically from DNA in the substitution of ribose for deoxyribose in the sugar backbone of the molecule, and of uridine (U) for thymine as one of the pyrimidine bases. Also, RNA normally exists as a single-stranded rather than double-stranded molecule. Recent advances have demonstrated far more diverse functions for RNA than were previously appreciated, particularly involving genes that produce functional RNA products that do not code for proteins. These probably represent at least 5 percent of all human genes, as suggested by current knowledge [Strachan and Read, 2010]. Several distinct classes of RNA molecules have been recognized, most of which are involved with regulating or assisting gene expression.
MicroRNA
MicroRNAs (miRNAs) are another class of small noncoding genes that regulate the expression of protein-encoding genes at the post-transcriptional RNA level [Denli et al., 2004]. The process begins with transcription (synthesis) of primary RNA transcripts that range in size from several hundred to several thousand kb. These transcripts are recognized and cut into precursor miRNAs in the nucleus by a protein known as Dicer, moved to the cytoplasm, and processed into mature miRNAs. The mature miRNAs join the RNA-induced silencing complex (RISC), which recognizes and cleaves (or otherwise silences) a target gene. This process has been demonstrated in many organisms, including mammals, and appears likely to play a key role in regulation of many genes.
Structure and Function of Polypeptides and Proteins
Proteins are composed of one or more polypeptide chains. Polypeptides are large polymers or macromolecules composed of linear sequences of repeating units known as amino acids, which are more complex than the repeating units of DNA or RNA. Amino acids consist of a three-carbon backbone, with an amino group attached to carbon 1 and a carboxyl group to carbon 3. They differ in the composition of a side chain attached to carbon 2. With rare exceptions, all polypeptides and proteins in nature are built from different sequences of 20 amino acids (Table 30-3). The side chains may be neutral and hydrophobic, neutral and polar, basic, or acidic. The simplest amino acid is valine, which has a hydrogen ion as the side chain.
Table 30-3 Classification of Amino Acids by Side Chain
| Amino Acid | 3-letter Code | 1-letter Code |
|---|---|---|
| Neutral and Hydrophobic | ||
| Alanine | Ala | A |
| Isoleucine | Ile | I |
| Leucine | Leu | L |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Proline | Pro | P |
| Tryptophan | Trp | W |
| Valine | Val | V |
| Neutral and Polar | ||
| Asparagine | Asn | N |
| Cysteine | Cys | C |
| Glutamine | Glu | Q |
| Glycine | Gly | G |
| Serine | Ser | S |
| Threonine | Thr | T |
| Tyrosine | Tyr | Y |
| Acidic | ||
| Aspartic acid | Asp | D |
| Glutamic acid | Glu | E |
| Basic | ||
| Arginine | Arg | R |
| Histidine | His | H |
| Lysine | Lys | K |
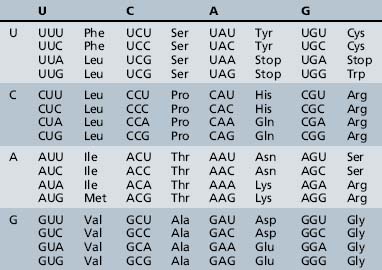
The process of information transfer from RNA polypeptides to proteins is known as translation. It relies on the genetic code, the system by which the nucleotide sequence of mRNA specifies the amino acid sequence of a polypeptide chain. In this nearly universal code, each set of three adjacent bases in the mRNA transcript constitutes a codon, and different combinations of bases within the codon specify the individual amino acids (Table 30-4). The small tRNA molecules serve as the molecular link between mRNA codons and amino acids. One segment of each tRNA transcript contains a three-base anticodon that is complementary to a specific codon on the mRNA, whereas another segment contains a binding site for one of the 20 amino acids.
Gene Structure and Organization
As noted earlier, a gene traditionally has been defined as a unit of genetic information. This concept has gradually progressed to a more useful definition, which states that a gene is a sequence of DNA on a chromosome that is required for production of a functional product, which can be either a protein or a functional RNA molecule [Nussbaum et al., 2007]. By convention, genetic information is always read in the 5′ to 3′ direction, whether encoded in DNA or RNA – in an upstream to downstream direction. The nomenclature regarding the 5′ and 3′ positions of the sugar backbone can be confusing. The 5′ carbon of the first nucleotide of a sequence is joined by a phosphodiester bond to a nucleotide not involved in the sequence, whereas its 3′ carbon is joined to the 5′ carbon of the second nucleotide, and so on. The last nucleotide of the sequence has a 3′ carbon, which joins another uninvolved nucleotide.
Genes
A model of a typical human gene is shown in Figure 30-3. Promoter sequences required for regulation and initiation of RNA transcription (red diamonds in Figure 30-3) are present at the 5′ end of the gene, such as the CAT and TATA boxes whose sequences are tightly conserved among many different genes and species. Downstream from the promoter sequences is a specific sequence that signals the start of transcription. A short way further downstream is an initiator codon, AUG, which codes for methionine. This triplet is the translation start site, which signals the start of the coding sequence for the polypeptide product. The region between the transcription and translation start sites is the 5′ UTR.
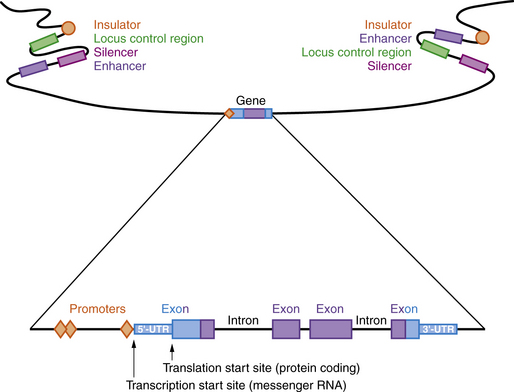
Regulatory Regions
Many genes have highly conserved sequences, a longer distance upstream and downstream of the transcribed gene, that are involved in regulating expression, including enhancers, silencers, locus control regions, and insulators (see Figure 30-3). Enhancer elements function to increase gene expression, while silencers reduce gene expression. Locus control regions may regulate expression of several genes within a chromosome region, while insulators prevent co-regulation of more distant genes and gene regions. All of these are sequences that bind proteins called transcription factors, which can be ubiquitous, tissue-specific, and/or temporally expressed. Promoters are located immediately 5′ of the gene and bind to RNA polymerase II, a necessary step for transcription. Other transcription factors bind upstream of the promoter and activate transcription. Enhancers and silencers are often located at a distance from the promoter, and increase or decrease transcription in a tissue-specific or temporal manner. Overall, the transcription of each gene is tightly regulated, with multiple transcription factors involved.
RNA Processing
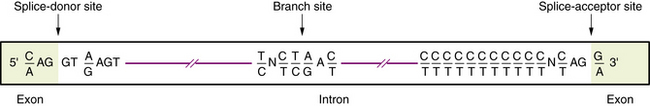
Transcription of DNA gives rise to a precursor RNA that corresponds exactly to the genome sequence but must be modified in several ways to become functional, especially for mRNA. The first modification to mRNA is the addition of a CAP structure to the 5′ end and this is followed by the removal or splicing of introns. The mechanism of mRNA splicing depends on the specific nucleotide sequences at the exon/intron boundaries called splice junctions (Figure 30-4). The most important of these is the GT-AG rule: introns almost always start with GT (actually GU, because this occurs in RNA), which is therefore called the splice-donor site, and end with AG, which is called the splice-acceptor site. Several additional specific sequences are also needed, including sequences within the intron just after the GT splice-donor site, at a highly conserved branch site located about 40 bp before the end of the intron and just before the AG splice-acceptor site. The splicing mechanism produces the following:
Imprinting and X-Inactivation
Imprinting
The process by which certain genes in specific chromosomal regions are expressed from only one chromosome, depending on the parental origin of the chromosome, is known as “imprinting.” Although the mechanism is only partly understood, a key component involves allele-specific DNA methylation, found predominantly at the carbon 5 position of about 80 percent of all cytosines that are part of symmetrical cytosine-guanine (CpG) dinucleotides [Jiang et al., 2004; Strachan and Read, 2010; Weksberg et al., 2003].
This process is controlled by regulatory imprinting “centers,” located nearby on the same chromosome as that of the silenced or “imprinted” gene. In effect, then, two alleles of the same gene that are identical in nucleotide sequence but derived from opposite parents are regulated differently in the same nucleus. This process is reversible, so that the silent, imprinted allele can be reactivated and the active allele silenced when passed through the germline of the opposite-sex parent. Most imprinted genes are found in large clusters of greater than 1 Mb (megabase pairs) in length. Imprinted clusters have been identified in chromosomes 6q24, 7p11.2, 11p15.5, 14q32, 15q11–q13, and 20q13.2, and others may exist as well [Cavaille et al., 2002; Gardner et al., 2000; Hall, 1990; Jiang et al., 2004; Weksberg et al., 2003; Wylie et al., 2000]. Imprinted regions share several common characteristics, including differential DNA methylation, allele-specific RNA transcription, antisense transcripts, histone modifications, and differences in timing of replication.
X-Inactivation
In mammalian cells with two (or more) X chromosomes, all but one undergo widespread gene silencing by methylation. This phenomenon, known as X-chromosome inactivation (Xi), causes one of the two X chromosomes in cells of female mammals to become transcriptionally inactive early in embryonic development, a phenomenon known as the Lyon hypothesis [Lyon, 1961, 2002]. In mutant cells with more than two X chromosomes, all but one become inactivated. This has the effect of balancing gene dosage of X-linked genes between male and female cells. The process of Xi is random, so that on average the maternally and paternally derived X chromosomes are each inactivated in approximately 50 percent of cells. Changes in this pattern are seen in female carriers of some X-linked diseases, resulting in skewing of Xi. This alteration can be favorable, with decreased severity of the phenotype, or unfavorable, with increased severity of the phenotype [Dobyns et al., 2004].
Cell Cycle and Chromosomal Basis of Heredity
Cell Cycle
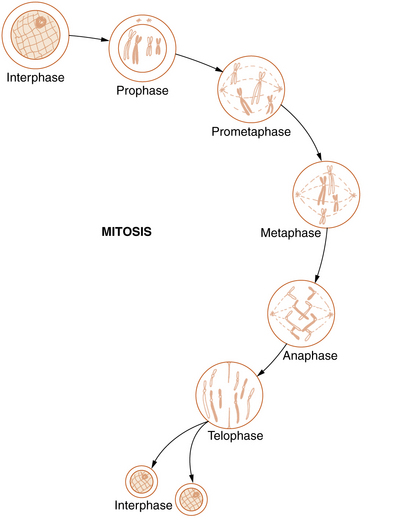
Mitosis
Somatic cell division, or mitosis, is an elaborate mechanism that distributes one chromatid of each duplicated chromosome to each of the two daughter cells. The process is continuous but has been divided into the following five stages: prophase, prometaphase, metaphase, anaphase, and telophase (Figure 30-5).
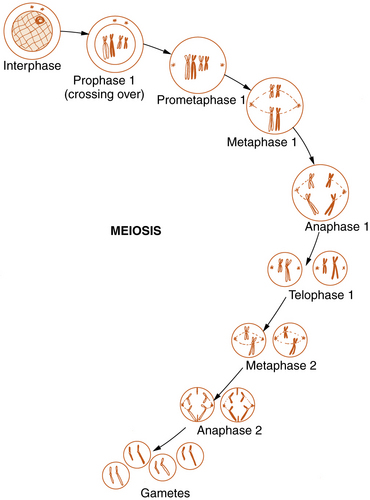
Meiosis
Reproductive cell division, or meiosis, is an even more complex mechanism in which two successive cell divisions, known as meiosis 1 and meiosis 2, give rise to the haploid germ cells (Figure 30-6). Meiosis is of critical importance in understanding many of the methods of modern molecular genetics and the pathogenesis of many genetic diseases.
Chromosomal Basis of Heredity
Chromosome Structure
In humans, the nuclear DNA is dispersed among 46 separate linear structures or chromosomes, each of which consists of a single, uninterrupted double helix that contains 50–250Mb of DNA, and a group of associated proteins that form the support structure or scaffolding. The scaffolding consists of five basic proteins called histones and several more acidic nonhistone proteins. Two copies of each of four histones – H2A, H2B, H3, and H4 – join to form an octamer. The DNA double helix wraps almost twice around the octamer, which involves about 140 bp. Adjacent octamers are separated by a short spacer segment of 20–60bp that is associated with histone H1. The complex of DNA and core histones is known as a nucleosome (see Figure 30-2).
Strings of nucleosomes are further compacted into a secondary helical structure known as a solenoid. These structures have a diameter of about 30 nm (see Figure 30-2) and contain six nucleosomes per turn. The solenoids are packed into large loops of 10–100 kb of DNA, which are attached to a nonhistone protein scaffolding. These loops pack together loosely to form interphase chromosomes. During early prophase, they pack together more closely to form knoblike thickenings known as chromomeres, which then coalesce further to form the bands observed in prometaphase and metaphase chromosomes when stained with appropriate dyes.
Specialized Regions
Origins of replication are specialized sequences where DNA replication begins, and thus are important in maintaining chromosome number and integrity. They consist of autonomously replicating sequence elements that contain a core consensus sequence and some imperfect copies with a length of about 50 nucleotides. A consensus human autonomously replicating sequence has been identified [Strachan and Read, 2010].
Chromosome Number
Each human somatic cell contains 46 chromosomes that consist of 22 matched pairs known as autosomes and two sex chromosomes: XX in females and XY in males (Figure 30-7). In contrast, human germ cells contain only 23 chromosomes, consisting of 22 unpaired autosomes and a single sex chromosome. The former is known as the diploid or 2n number, and the latter is known as the haploid or 1n number. The autosomes were numbered according to length, with chromosome 1 the longest and chromosome 22 thought to be the shortest. Although chromosome 21 later proved to be shorter than chromosome 22, the numbers were retained for historical reasons. The two members of each pair of autosomes and the two X chromosomes in females carry the same genes and are known as homologous chromosomes, or homologs. Although they appear similar under the microscope, homologs are not strictly identical. They contain the same genes, but the nucleotide sequence differs at thousands of positions.

Organization of the Human Genome
The human genome comprises the total of all genetic information in the cell. It is divided into two separate compartments – a large and complex nuclear genome and a much smaller and simpler mitochondrial genome. The mitochondrial genome consists of a single circular DNA molecule that is present in many copies in each mitochondrion, while the nuclear genome is distributed among the 46 nuclear chromosomes. The available data regarding the genome have become much more extensive and accurate with completion of the Human Genome Project. A few of the most useful Human Genome Project-related websites are listed in Table 30-1.
The Nuclear Genome
The human nuclear genome consists of approximately 3 × 109 bp, or 3000 Mb of DNA. About 75 percent of this represents unique or single-copy DNA, which includes genes and some important regulatory elements. The remaining 25 percent consists of several classes of repetitive DNA [Lander et al., 2001; Nussbaum et al., 2007; Venter et al., 2001].
Genes and Conserved Noncoding DNA
Somewhat surprisingly, recent estimates predict that the human genome contains less than 30,000 protein-coding genes (possibly closer to 20,000) and an uncertain number of other genes producing functional RNA products. This is far fewer than earlier estimates, and accounts for only about 1.2 percent of nuclear DNA [Lander et al., 2001; Venter et al., 2001]. Another 5 percent of the human genome is more conserved than would be expected from estimates of neutral evolution, which suggests that many of these regions have specific, regulatory functions [Chiaromonte et al., 2003; Waterston et al., 2002]. Studies of these highly conserved regions of DNA have used different thresholds, such as stretches of more than 100 bp with 70–80 percent conservation between mouse and human. Some of these regions have been found to contain important noncoding elements [Dermitzakis et al., 2002, 2003; Frazer et al., 2004; Hardison, 2000]. More stringent analysis demonstrates that the human genome contains 481 sequences of 200 or more bp that are 100 percent conserved among human, mouse, and rat [Bejerano et al., 2004]. These segments were designated “ultra-conserved elements,” and are preferentially located near genes involved in RNA processing or regulation of transcription and development. Similarly, about 5000 sequences of 100 bp or more are conserved among these three species, which emphasizes that noncoding sequences are common and important.
Low Copy Repeats
Segmental duplications, also known as low copy repeats (LCRs), are DNA sequences of 10–250 kb, present in multiple copies with greater than 95 percent sequence identity, that make up approximately 5 percent of the human genome [Babcock et al., 2003; Bailey et al., 2002; Cheung et al., 2001; Stankiewicz and Lupski, 2002]. LCRs are dynamic regions of the genome because specific repeats tend to cluster within the same genomic regions, where they mediate unequal nonhomologous recombination events, producing segmental deletions and duplications that are collectively designated “copy number variants” (CNVs). Several of these have been associated with well-known developmental disorders in humans, such as Williams’ syndrome in 7q11.23, Angelman’s syndrome and Prader–Willi syndrome in 15q12, hereditary neuropathy with predisposition to pressure palsies and Charcot–Marie–Tooth neuropathy type 1A in 17p12, Smith–Magenis syndrome in 17p11.2, and DiGeorge’s syndrome in 22q11.2 [Babcock et al., 2003]. Many new CNV-associated devlopmental brain disorders have been described over the past few years.
Polymorphisms
A mutation is a permanent change in the DNA of an individual organism, specifically a change in the nucleotide sequence anywhere in the genome [Nussbaum et al., 2007]. Genetic diseases and many cancers are caused by mutations that adversely affect function of one or more genes, although most mutations have little or no effect on gene function and therefore do not change the survival or reproductive fitness of an individual. Some of these persist in the population as morphologic variants known as polymorphisms. Sequence changes that have frequencies of less than 1 percent are known as rare variants, whereas those with frequencies of 1 percent or more are known as polymorphisms. By convention, a genetic polymorphism is defined as the occurrence of two or more variants or alleles in a region of DNA where at least two alleles appear with frequencies greater than 1 percent. Several different classes of polymorphisms occur in the genome, and several methods in molecular biology take advantage of the normal variation between individuals.
Microsatellites
The most common microsatellite family consists of 50,000–100,000 cytosine-adenine (CA) repeats, which consist of short tandem repeats of the dinucleotide CA on one strand and guanine-thymine on the complementary strand. They thus take the form (CA)n/(GT)n, with n in the range of 6–30 [Weber and May, 1989]. The number of repeats within a (CA)n block varies greatly among different members of a species, producing a set of alleles that always differ in size by multiples of two bases. About 70 percent of the human population is heterozygous at any given (CA)n repeat locus, making these highly polymorphic. The human genome contains about 50,000–100,000 interspersed (CA)n blocks, which is enough to place 1 block every 30–60 kb, if evenly spaced.
Single-Nucleotide Polymorphisms
Single-nucleotide polymorphisms (SNPs, pronounced “snips”) are DNA sequence variations that occur when a single nucleotide (A, T, C, or G) in the genome sequence is changed. For example, a SNP might change the DNA sequence TCACG to TTACG. The most common sequence change involves replacement of cytosine (C) with thymidine (T), which accounts for about two-thirds of all SNPs. As with other types of sequence variation, a SNP must occur in at least 1 percent of the population to be classified as a polymorphism. SNPs occur in both unique-sequence (coding and noncoding) and repetitive DNA, and are responsible for about 90 percent of human genetic variation. On average, SNPs are found approximately every 100–300 bases along the entire human genome. Although most SNPs likely have no function, some are known to influence disease predisposition or responses to drugs, and thus are proving to be very valuable in studying the causes of common human diseases. The current inventory of known SNPs can be found in the Human SNP database (dbSNP) on the NCBI Entrez website (see Table 30-1).
Mitochondrial Genome
Mitochondria are cellular organelles that are primarily responsible for cellular respiration and production of adenosine triphosphate. Each cell contains numerous mitochondria, and each mitochondrion contains many copies of a small 16.5-kb circular chromosome, adding up to thousands per cell. The mitochondrial chromosome contains 37 genes that code for two types of rRNA, 22 types of tRNA, and 13 polypeptides. The two DNA strands differ significantly in base composition, with a heavy strand rich in guanines that codes for 28 genes, and a light strand rich in cytosines that codes for 9 genes. It is very densely packed, with 93 percent comprising coding sequence [Strachan and Read, 2010].
Human Genome Project
The importance of DNA, including both genes and noncoding regions, became increasingly apparent during the 1970s and 1980s, leading to one of the most ambitious scientific research projects ever undertaken – a plan to sequence the entire human genome. This project, which was begun in 1990, came to be known as the Human Genome Project. The goals of the project, as taken from the Human Genome Project website (see Table 30-1), were as follows:
< div class='tao-gold-member'>


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


