Chapter 23 Disorders of Forebrain Development
Introduction
The prosencephalon forms at the end of primary neurulation as one of three principal vesicles: the hindbrain, the midbrain, and the forebrain (prosencephalon). Detailed embryology of the normal prosencephalon is beyond the scope of this chapter, but is covered comprehensively in Rao and Jacobson [2005]. The primary disorders of prosencephalic formation, prosencephaly and atelencephaly, are extremely rare and not further discussed in this chapter. The major disorders of prosencephalic formation, holoprosencephaly, agenesis of the corpus callosum, and septo-optic dysplasia, are discussed below, after a brief introduction to prosencephalic development.
Prosencephalon Patterning
Prosencephalic development occurs by inductive interactions under the primary influence of the prechordal mesoderm. The peak time period of development is the second and third months of gestation, with the earliest prominent phases in the fifth and sixth weeks of gestation. The major inductive relationship of concern is between the notochord/prechordal mesoderm and the forebrain. This interaction occurs ventrally at the rostral end of the embryo, and thus the term ventral induction is sometimes used. The inductive interaction influences formation of much of the face and of the forebrain; accordingly, severe disorders of brain development at this time usually result in striking facial anomalies. Development of the prosencephalon is considered best in terms of three sequential events: prosencephalic formation, prosencephalic cleavage, and midline prosencephalic development (Figure 23-1). Prosencephalic formation begins at the rostral end of the neural tube at the end of the first month and the beginning of the second month of gestation. It consists of segmentation of the prosencephalon into three major domains, termed prosomeres (P1–P3). P1 gives rise to the pretectum, the region of brain immediately rostral to the midbrain-derived tectum. P2 is associated with development of the thalamus, and P3 gives rise to the prethalamus. More rostral brain regions, including the telencephalon, are also divided into prosomeric boundaries, the clearest of which are the anatomic boundaries of the developing basal ganglia, the medial and lateral basal ganglia, which arise as bulges along the ventrolateral telencephalon and express the markers Dlx and Arx. The neocortex itself exhibits regionally restricted gene expression. Manipulation of these expression patterns results in shifting of cellular identities of the neocortex (FGF8, EMX2). The evidence to date, however, argues against anatomically and regionally restricted boundaries, because cell lineage experiments demonstrate that sibling cells can occupy multiple nuclei throughout the anteroposterior axis.
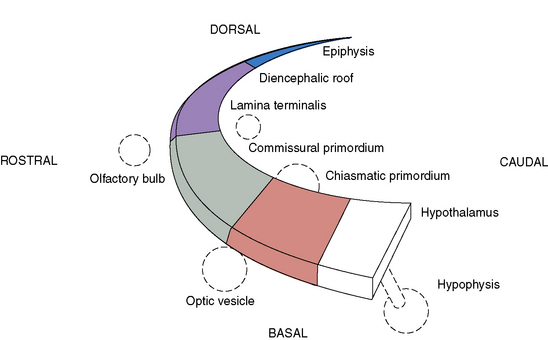
Prosencephalic Cleavage
Three crucial thickenings or plates of tissue become apparent around the end of the second month; these are the commissural, the chiasmatic, and the hypothalamic plates. These structures are important in the formation, respectively, of the corpus callosum and septum pellucidum, the optic nerve chiasm, and the hypothalamic structures. The most prominent of these embryologic changes is the formation of the corpus callosum, the earliest component of which appears at 9 weeks. Disorders associated with abnormal development of the prosencephalon are outlined in Table 23-1.
Table 23-1 Disorders of Midline Prosencephalic Development
| Region Affected | Disorder |
|---|---|
| Commissural plate | Agenesis of the corpus callosum and/or septum pellucidum |
| Commissural and chiasmatic plates | Septo-optic dysplasia |
| Commissural, chiasmatic, and hypothalamic plates | Septo-optic-hypothalamic dysplasia |
Holoprosencephaly
Holoprosencephaly (HPE) is a complex brain malformation characterized by a failure of the forebrain (prosencephalon) to separate completely into two distinct cerebral hemispheres (i.e., distinct telencephalon and diencephalon). This process is normally completed by the fifth week of gestation [Golden, 1999].
HPE is typically associated with midline facial anomalies.
Historical Background
Individuals with cyclopia have been described for centuries in mythology and in the scientific literature since the late 18th century [Siebert et al., 1990]. In 1882, Kundrat first described the cerebral changes of HPE, including absent olfactory nerves. Believing the absence of olfactory lobes and bulbs was the cardinal feature, he termed the condition “arhinencephaly” [Kundrat, 1882]. Yakovlev recognized involvement of the entire telencephalon and called the single telencephalic ventricle a “holosphere” and the malformation “holotelencephaly” [Yakovlev, 1959]. DeMyer found that the thalamus and other diencephalic structures were also involved and coined the still-favored term “holoprosencephaly” to indicate that the defect involved the entire prosencephalon [DeMyer and Zeman, 1963].
Epidemiology
HPE is the most common developmental defect of the forebrain and midface in humans, and occurs in 1 in 250 pregnancies [Matsunaga and Shiota, 1977], but because only 3 percent of the fetuses with HPE survive to delivery, the incidence in live births is only approximately 1:10,000 [Bullen et al., 2001; Croen et al., 2000; Forrester and Merz, 2000; Rasmussen et al., 1996]. Two-thirds of affected patients have the most severe subtype of HPE [Ming and Muenke, 1998]. However, high-resolution magnetic resonance imaging (MRI) has increased identification of children with less severe forms who are not diagnosed until later in infancy. Therefore, the true live birth prevalence of HPE may be higher than previously estimated, and the proportion of cases with milder subtypes appears to be increasing in more recently reported series [Stashinko et al., 2004]. There also appears to be a slight female preponderance in some case series. A few studies of limited size suggest a higher than average prevalence of HPE in Far East Asians and Filipinos [Forrester and Merz, 2000].
Definition and Subtypes of Holoprosencephaly
The sine qua non of HPE is an incomplete separation of the cerebral hemispheres that results in lack of cleavage (nonseparation) of midline structures involving the telencephalon and diencephalon. HPE typically is divided into three main subtypes, delineated by DeMyer and co-workers [DeMyer, 1987; DeMyer and Zeman, 1963] and distinguished by the degree of separation of the cerebral hemispheres (Figure 23-2).
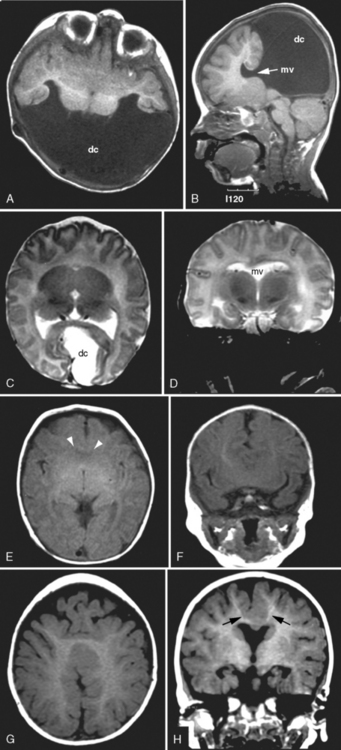
Fig. 23-2 Subtypes of holoprosencephaly.
(From Hahn JS, Plawner LL. Evaluation and management of children with holoprosencephaly. Pediatr Neurol 2004;31:79.)
In addition to DeMyer’s classic HPE types, another subtype is identifiable, the middle interhemispheric variant [Simon et al., 2002]. In this variant, the midportion of the cerebral hemispheres is continuous across the midline, with absence of the corpus callosum seen only in this region. There is separation of the anterior frontal lobes, basal forebrain, and occipital lobes. Evidence for this malformation being a subtype of HPE is bolstered by the finding that a mutation in the ZIC2 gene, which has been implicated in causing the classic forms of HPE, also has been found in patients with the middle interhemispheric variant [Brown et al., 2001; Solomon et al., 2010].
In addition to nonseparation of the cerebral hemispheres, failure of separation also is common in the hypothalamic, caudate, lentiform, and thalamic nuclei. About one-quarter of patients have some degree of midbrain nonseparation [Simon et al., 2000]. Occasionally, isolated neuronal heterotopias are seen, particularly in the middle interhemispheric variant. The gyri often are normally developed, although in alobar and semilobar HPE the gyri may be excessively smooth or broad [Barkovich et al., 2002].
Although HPE is typically divided into these subtypes, the degree of malformation and brain regions involved occur along a spectrum, and individual patients frequently do not fall neatly into any one category [Hahn and Barnes, 2010]. Milder cases are being increasingly identified with use of high-resolution MRI. For example, a mild form of lobar HPE (septo-preoptic type) can be recognized, with minimal cortical fusion restricted to the preoptic regions (involving the suprachiasmic region and anterior hypothalamus) and/or the septal region (subcallosal region) [Hahn et al., 2010].
Neuropathological Findings
Cytoarchitecturally, the brain usually shows normal cortical layering; in some rare cases, the histopathology may show disorganization or abnormal lamination [Golden, 1999]. The defects in cortical organization may represent secondary injury to the cerebral cortex or an abnormality in connections into and out of the cerebral cortex. Periventricular and white-matter glioneuronal heterotopia are also encountered in rare cases. The hippocampus is virtually always present, although it may show incomplete or abnormal development [Golden, 1999]. The cerebellum may show various degrees of cell migration abnormalities, such as misplaced nodules that are aberrantly located in the white matter or within the dentate nuclei [Larroche, 1977].
Etiology
Multiple environmental and genetic factors have been implicated in causing HPE. Prenatal exposures to a variety of toxins, medications, and infections also have been reported. Anecdotal reports suggest viruses, low-calorie diets, hypocholesterolemia, maternal diabetes, and use of salicylates, alcohol, and contraceptives as possible causes [Johnson and Rasmussen, 2010]. The strongest teratogenic evidence exists for maternal diabetes [Barr et al., 1983] and exposure to alcohol [Sulik and Johnston, 1982] and retinoic acid [Croen et al., 2000]. A diabetic mother’s risk of having a child with HPE is approximately 1 percent, a greater than 100-fold increase over the general population. A recent population study confirmed the risks of pre-existing maternal diabetes and salicylates (aspirin), but also noted an increased risk with artificial reproductive therapy [Miller et al., 2010]. Some teratogens are thought to produce HPE via interference with the sonic hedgehog gene signaling pathways, mediated by perturbations of either cholesterol biosynthesis or the ability of target tissue to sense or transduce the sonic hedgehog signal [Cohen and Shiota, 2002].
Approximately 30–50 percent of live births with HPE have chromosomal abnormalities [Bullen et al., 2001; Croen et al., 1996], but this is likely an overestimation based on under-reporting of milder cases. HPE can be seen in association with trisomy 13, trisomy 18, or triploidy. HPE can also be seen in several malformation syndromes having normal karyotypes, such as Pallister–Hall syndrome, Rubinstein–Taybi syndrome, and Smith–Lemli–Opitz syndrome [Siebert et al., 1990]. An updated list of genetic disorders associated with HPE can be found on the Online Mendelian Inheritance in Man (OMIM) website (http://www.ncbi.nlm.nih.gov/omim).
In nonsyndromic and nonchromosomal HPE, multiple pedigrees have been described, manifesting various inheritance patterns, most commonly autosomal-dominant and autosomal-recessive inheritance. At least nine genes have been associated with HPE, including SHH (7q36), ZIC2 (13q32), SIX3 (2p21), TGIF (18p11.3), PATCHED-1 (9q22), GLI2 (2q14), DISP1 (1q24), NODAL (10q), and FOXH1 (8q24.3) [Roessler and Muenke, 2010]. Of these genes, the four most commonly affected (SHH, ZIC2, SIX3, and TGIF) account for only 25 percent of the cases of HPE with normal chromosomes [Dubourg et al., 2004; Roessler and Muenke, 2010] and approximately 5–10 percent of all HPE patients.
SHH was the first identified HPE-associated mutated gene and has been the most extensively studied [Belloni et al., 1996]. The Shh protein is a secreted intercellular signaling molecule involved in establishing cell fates at several points during development. Shh is expressed early in development in the ventral forebrain and is critical for ventral patterning of the developing neural tube. It also is expressed in the ectoderm of the frontonasal and maxillary processes. Disruption of Shh signaling in several animal models produces the brain and facial malformations seen in HPE.
The SHH signaling network is the common pathway through which multiple environmental and genetic influences interact to cause HPE. Several of the other HPE genes are important components of the SHH signaling network; PTCH is a receptor for SHH and GLI2 is a mediator of SHH target gene transcription. A well-studied example of environmental influences acting through the SHH signaling network is the association of HPE with various causes of hypocholesterolemia [Ming and Muenke, 2002]. SHH is modified by cholesterol, which is felt to be essential for its proper functioning. Maternal hypocholesterolemia has been implicated in causing HPE. The incidence of HPE is also increased in Smith–Lemli–Opitz syndrome, which is due to a defect in 7-dehydrocholesterol reductase, the final enzyme in cholesterol synthesis [Kelley et al., 1996]. In addition, an outbreak of cyclopia (severe midline facial malformation) in sheep was believed to be caused by ingestion of plants containing cyclopamine and jervine, compounds that interfere with sonic hedgehog signaling by binding to and thereby sequestering the protein “smoothened” [Keeler and Binns, 1968].
In addition to the SHH gene, the Nodal signaling pathway also appears to be implicated in HPE. This nodal pathway includes TGIF, NODAL, GDF1, TDGF1, and FOXH1. A complex genetic interaction involving these gene products and which causes a reduction in nodal signaling may produce HPE [Roessler and Muenke, 2010; Roessler et al., 2008].
Studies of genotype–phenotype correlation in mutations of SHH have found that the same mutation can result in extremely variable phenotypes. Even within the same family, an identical mutation in SHH may result in a severe brain malformation, a “mild sign” known as a microform (such as a single median maxillary central incisor), or no phenotypic abnormality. It is estimated that, among carriers of an abnormal gene, 37 percent will have HPE, 27 percent will have a microform, and 36 percent will have no clinical abnormality [Cohen, 1989]. This great phenotypic variability with the same genetic mutation has suggested that, although HPE was thought of as a single-gene disorder, it should be considered a multifactorial disorder, with the specific phenotype being the result of multiple genetic and environmental influences [Ming and Muenke, 2002]. A large majority of HPE cases, therefore, appear likely to be due to a disturbance of a complex network of interacting genes and signaling centers [Roessler and Muenke, 2010]. In addition, the timing of teratogenic exposure appears to be critical to the specific resulting phenotype. An experimental model using cyclopamine, an inhibitor of cholesterol synthesis, has revealed that varying the time of exposure resulted in a continuum of midline facial malformation [Cordero et al., 2004].
Clinical Manifestations and Outcomes
Along with the midline brain malformation seen in HPE, a corresponding midline facial malformation may be present. In its most severe and usually lethal form, cyclopia, with the presence of a single midline eye and a proboscis (rudimentary single-nostril nose) above the eye, can be present. Survivors may have hypotelorism, a flattened nasal bridge, median cleft lip and palate, or a single median maxillary central incisor. The oft-quoted statement “the face predicts the brain” [DeMyer et al., 1964] refers to the observation that the degree of facial malformation frequently reflects the degree of brain malformation. This was amended later to “the face predicts the brain approximately 80 percent of the time,” in recognition of individuals with alobar HPE with a normal facial appearance, as well as cases of milder brain malformation associated with abnormal facies [Cohen, 1989].
Previous studies indicated that children with HPE do not survive beyond early infancy. This may have been due to the identification of only the most severe cases. Early death is typical for most cytogenetically abnormal children and those individuals with the most severe facial features (cyclopia or ethmocephaly) [Croen et al., 1996]. In children without these risk factors, more recent studies have indicated that long-term survival is not uncommon. Among cytogenetically normal patients with all types of HPE, more than 50 percent are alive at 12 months [Barr and Cohen, 1999; Olsen et al., 1997]. In one series of 104 HPE patients, the mean age was 4 years, and 15 percent were between 10 and 19 years of age [Hahn and Plawner, 2004]. Even with the most severe type, alobar HPE, children may survive. In an observational study of alobar HPE, 30 percent were alive at 12 months, with long-term survival noted as well [Barr and Cohen, 1999]. When death did occur in the alobar group, causes were associated with brainstem dysfunction, pneumonia, dehydration from diabetes insipidus, and rarely, intractable seizures.
Children with HPE may experience a variety of medical and neurologic problems. A significant proportion develop hydrocephalus. The likelihood of requiring ventriculoperitoneal shunting is much higher in alobar than in semilobar or lobar HPE (60 percent versus 8 percent, respectively) [Plawner et al., 2002]. The risk of developing hydrocephalus also is higher when a dorsal cyst is present [Simon et al., 2001]. The dorsal cyst is thought to form as a result of the thalami being fused across the midline, thereby blocking cerebrospinal fluid egress from the third ventricle and disrupting cerebrospinal fluid. Most children that have HPE without hydrocephalus are microcephalic.
As in children with other midline brain defects, endocrinologic problems are very common. Diabetes insipidus is particularly frequent, and in one series of 68 children, three-quarters of individuals were affected [Plawner et al., 2002]. Diabetes insipidus can be insidious in presentation; serum sodium concentrations of 160 mEq/L or greater have been occasionally detected on routine electrolyte evaluation and were not accompanied by any acute symptoms. Diabetes insipidus also can have a fluctuating course. Growth hormone deficiency, hypocortisolism, and hypothyroidism also may occur. The endocrinopathies may be due to midline defects involving the hypothalamus and are rarely due to a dysgenetic (e.g., hypoplastic or ectopic) pituitary gland.
Approximately half of the children with HPE have epilepsy, and the likelihood of developing seizures does not correlate with the severity of the brain malformation. The most common seizure type is complex partial seizures, with or without secondary generalization. Seizure type, however, can be variable, including generalized tonic-clonic, tonic, atonic, myoclonic seizures or infantile spasms [Hahn and Plawner, 2004]. In 50 percent of affected children, the seizures are relatively easy to control with antiepileptic medication. Having a concurrent area of cortical dysplasia is a risk factor for having difficult-to-control seizures.
Feeding problems and swallowing dysfunction are common in children with HPE, and are correlated with the severity of the brain malformation. Two-thirds of patients with alobar and semilobar HPE require gastrostomy tubes [Plawner et al., 2002]. Abnormalities of muscle tone are also very common in HPE, and the severity correlates with the severity of the brain malformation. Many children have early hypotonia, followed by the development of upper limb and oromotor dystonia, and lower limb spasticity [Hahn and Plawner, 2004].
Developmental disability affects nearly all patients with HPE. The severity of the brain malformation determines the degree of delay and neurological impairments. Severe developmental delay is present in alobar HPE [Barr and Cohen, 1999]. There is no reported case of a child with alobar HPE who is able to sit independently. In lobar HPE, approximately 50 percent of children ambulate (with or without assistance), use their hands functionally, and have some verbal communication [Plawner et al., 2002]. Formal neuropsychologic evaluation in all types of HPE demonstrates relative strengths in receptive language and socialization, and weaknesses in visual reasoning and nonverbal problem skills [Kovar et al., 2001].
Management
Children with alobar HPE, a dorsal cyst, or normo- or macrocephaly should be closely observed for the development of hydrocephalus. Most experts recommend ventriculoperitoneal shunting for hydrocephalus, even in severe HPE, because failure to institute this measure will lead to progressive head enlargement and greater difficulty in caring for the child [Barr and Cohen, 1999; Hahn and Plawner, 2004]. Electrolyte screening should be performed to assess for the subacute development of diabetes insipidus. Screening for other endocrine abnormalities should be considered less frequently, with assays of cortisol, thyroid-stimulating hormone, free thyroxine, and insulin-like growth factor-1 (IGF-1). Because seizures may not develop in half of the children with HPE, prophylactic antiepileptic drugs are not recommended. If seizures do develop, the possibility of acute reactive seizures should be evaluated for and a serum sodium level checked. MRI also should be considered to evaluate for focal heterotopia. Gastrostomy tubes often are necessary to address the complex management issues of ensuring adequate calories related to feeding dysfunction and delivery of adequate free water necessary for management of diabetes insipidus. Motor difficulties and dystonia may be partially responsive to trihexyphenidyl. This agent may improve upper extremity and oromotor function [Hahn and Plawner, 2004]. Motor dysfunction, including hypertonia and dystonia, is common and may require physical, pharmacological, or surgical therapies.
Prenatal Diagnosis and Imaging
The sensitivity of first-trimester ultrasound examination in the detection of HPE remains unclear. It can detect alobar HPE [Filly et al., 1984; Sepulveda et al., 2004], but may be much less sensitive in detecting milder cases. A recent study from Germany reported a series of 51 fetuses (79 percent having a chromosomal abnormality) diagnosed with HPE by ultrasound at a mean age of 22 weeks [Wenghoefer et al., 2010]. The presence of large dorsal cysts, hydrocephalus, or midline craniofacial defects may provide clues that eventually lead to the recognition of the associated HPE.
Fetal MRI has been used to diagnosis various forms of HPE, including alobar, semilobar, lobar [Dill et al., 2009; Wong et al., 2005], and middle interhemispheric variants [Pulitzer et al., 2004] (Figure 23-3). Other midline anomalies, such as agenesis of the corpus callosum (isolated or with interhemispheric cysts), absence of the septum pellucidum, and hydrocephalus with communication of the lateral ventricles, are sometimes misdiagnosed prenatally as HPE [Malinger et al., 2005].
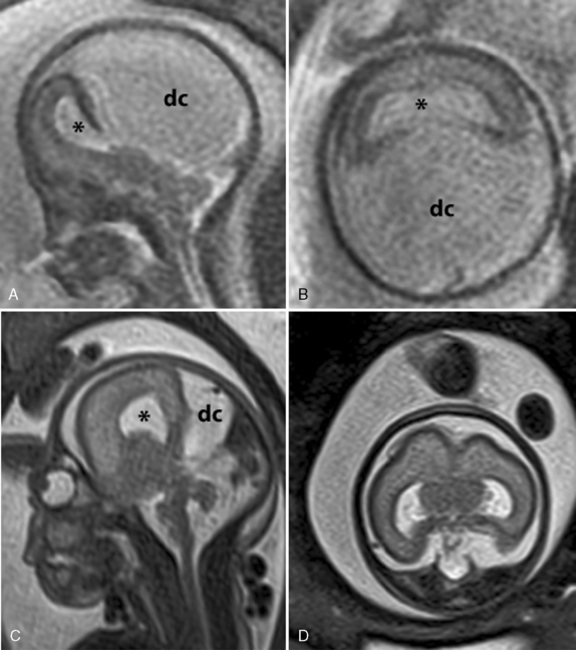
 -week gestational age fetus with alobar HPE. The thalami, basal ganglia, and midbrain structures are incompletely delineated on the midline sagittal image (A). A large dorsal cyst (dc) communicates with the monoventricle (asterisk). The hemispheres are not separated, resulting in a holosphere (B). C and D, Fetal MRI of a 26-week gestational age fetus with trisomy 13 and semilobar HPE. The HASTE fetal sequence in the midsagittal plane (C) shows a monoventricle (asterisk), a moderate size dorsal cyst (dc), and inferior cerebellar vermis hypoplasia (Dandy–Walker complex). On the axial image (D), the thalami and basal ganglia appear fused, while the posterior hemispheres are separated.
-week gestational age fetus with alobar HPE. The thalami, basal ganglia, and midbrain structures are incompletely delineated on the midline sagittal image (A). A large dorsal cyst (dc) communicates with the monoventricle (asterisk). The hemispheres are not separated, resulting in a holosphere (B). C and D, Fetal MRI of a 26-week gestational age fetus with trisomy 13 and semilobar HPE. The HASTE fetal sequence in the midsagittal plane (C) shows a monoventricle (asterisk), a moderate size dorsal cyst (dc), and inferior cerebellar vermis hypoplasia (Dandy–Walker complex). On the axial image (D), the thalami and basal ganglia appear fused, while the posterior hemispheres are separated.Genetic Counseling and Testing
The recurrence risk of isolated HPE is estimated to be 6 percent [Roach et al., 1975]. Special attention should be given to the family history, to identify “microforms,” such as anosmia or a single central incisor. Such a finding would indicate a substantially higher recurrence risk. Genetic testing for SIX3, SHH, TGIF, and ZIC2 is commercially available and should be considered if familial HPE is suspected. Prenatal testing for these genes is possible by means of amniocentesis or chorionic villus sampling. The gene tests, in conjunction with fetal MRI, have been found to be helpful in prenatal diagnosis and counseling in a series of pregnancies [Mercier et al., 2010]. High-resolution cytogenetic analysis is important in every fetus or newborn with HPE, since abnormalities can be identified in 24–45 percent of all individuals. Newer molecular methods, including subtelomeric multiplex ligation-dependent probe amplification and comparative genomic hybridization microarray testing, have identified chromosomal deletions, duplications, and unbalanced rearrangements in some patients with HPE. These tests may be useful as an addition to the chromosomal and HPE gene mutational analyses [Pineda-Alvarez et al., 2010].
Agenesis of the Corpus Callosum
The corpus callosum forms between the 8th and 14th weeks of fetal development. Nearly 200 million axons course through this structure and innervate each opposing hemisphere in a homotopic manner; that is, each axon innervates structures in the mirror location in the opposing hemisphere. Absence or diminution of this structure is found in over 1 in 3000 live births, and is the most common birth defect of the central nervous system after spina bifida [Glass et al., 2008]. Callosal abnormalities are frequently seen with other malformations of brain development, including polymicrogyria, heterotopia, and midbrain and hindbrain abnormalities, but can present in an isolated manner (Figure 23-4). Many cases are also associated with birth defects in other organ systems, including ophthalmologic, cardiac, and renal defects. Agenesis of the corpus callosum (ACC) can be caused by chromosomal disorders, and there is increasing recognition that subtle copy number variants (CNVs) of particular genes may play a critical role in the etiology of ACC [Sherr et al., 2005; O’Driscoll et al., 2010]. Single-gene mutations and metabolic disorders can also disrupt callosal development. The best-known and most studied environmental cause of ACC, fetal alcohol syndrome, can also result in a significant reduction in white-matter volume. There is a great diversity of clinical outcomes for patients with ACC. Many individuals with ACC have deficits in social cognition, even with a normal IQ. Many of these individuals carry clinical diagnoses that place their phenotype on the autism spectrum. Conversely, individuals with normal IQ and social deficits are often found on MRI to have isolated ACC; no other brain malformations are found. In contrast, in children with ACC and associated brain anomalies, many have seizures and significant developmental impairment, including intellectual disabilities and cerebral palsy. In some individuals with ACC due to chromosomal, metabolic, or single-gene disorders, the outcome can be quite severe, including a significantly shortened life expectancy.
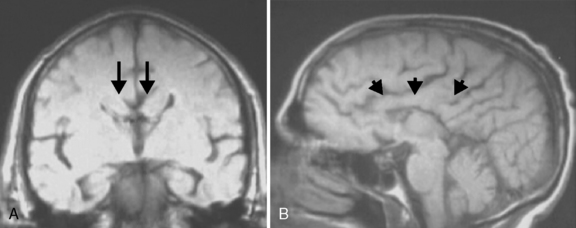
Fig. 23-4 Agenesis of the corpus callosum without other associated central nervous system malformations.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


