Investigational Drugs: Brivaracetam, Carisbamate, Eslicarbazepine Fluorofelbamate, Gamaxolone, Isovaleramide, Lacosamide (Harkoseride; SPM927), Losigamine, Retigabine, Safinamide, Seletracetam, Stiripentol, Talampanel, and Valrocemide
H. Steve White
Emilio Perucca
Michael D. Privitera
Introduction
During the 16 years between 1989 and 2005, a number of new antiepileptic drugs (AEDs) entered the market for the add-on treatment of partial seizures as their initially approved indication. The availability of these AEDs has contributed to the management of epilepsy, either as adjunctive therapy or as monotherapy. In addition to providing more choices for the physician and his/her patients, many of these drugs have led to improved seizure control and have been shown to possess a more favorable tolerability and pharmacokinetic profile than some of the older “first-generation” AEDs. Unfortunately, for the patient with refractory epilepsy, there still remains a substantial need for more efficacious therapies. Thus, none of the AEDs currently available can claim to be capable of providing complete seizure control in a sizeable proportion of patients with refractory partial epilepsy. In addition, only small advances in the therapy of certain seizure types and epilepsy syndromes have been made. Therefore, there is still significant room for improvement.
Despite the advancements in our understanding of the pathophysiology and molecular genetics of various epilepsies, little progress has been made in the development of a therapy that targets the presumed molecular basis of a given seizure type. To this end, several of the drugs in development today evolved from a nonmechanistic approach and the use of a battery of animal seizure and epilepsy models to define their in vivo anticonvulsant profile. This is not to imply that target-driven discovery is not employed in the discovery of investigational AEDs. As reviewed here, a number of the drugs in development have emerged from very focused programs designed around a “validated” molecular target.
Many of the drugs reviewed in this chapter will likely emerge from clinical studies over the next 2 to 5 years, and at least some of these new agents will offer some level of improvement to the multitude of patients whose seizures are not fully controlled with existing anticonvulsants. Whether any of these drugs will lead to complete seizure freedom in a major proportion of refractory patients or emerge ultimately as first-line agents for the treatment of one or more types of epilepsy has yet to be determined.
Brivaracetam (UCB 34714) and Seletracetam (UCB 44212)
Structure and Chemistry
Brivaracetam, (2S)-2-[(4R)-2-oxo-4-propylpyrolidinyl]bu-tanamide, and seletracetam, (2S)-2-[(4S)-4-(2,2,difluorovinyl)-2-oxopyrrolidin-1-yl] butanamide, are two very potent levetiracetam analogs in clinical development by UCB Pharma (Fig. 1).
Pharmacology
The development of brivaracetam and seletracetam stems from the recognition that the action of levetiracetam is related to its ability to bind with a novel site identified as synaptic vesicle protein 2A (SV2A).95 The demonstration that the efficacy of levetiracetam and a series of analogs is correlated with their SV2A binding affinity stimulated a drug-discovery program at UCB to identify compounds with higher affinity for SV2A than levetiracetam. Seletracetam and brivaracetam emerged from this effort, wherein approximately 12,000 compounds were initially screened against SV2A.64 These two analogs were chosen from 900 that were selected for in vivo evaluation in the audiogenic seizure-susceptible mouse, and from 30 that were subsequently selected for more extensive evaluation in the kindling model.
Seletracetam appears to be a more potent analog that shares a similar anticonvulsant profile with levetiracetam. For example, seletracetam is not active in the traditional maximal electroshock (MES) or pentylenetetrazole (PTZ) seizure models but is more potent than levetiracetam in the corneal-kindled mouse (10-fold) and the genetic absence epilepsy rat from Strasbourg (GAERS) model of spike-and-wave epilepsy (25-fold). In addition to increased potency, seletracetam also appears to possess an even higher protective index (i.e., the ratio of a drug’s median toxic dose to its median effective dose) in animal models than does levetiracetam.104
 FIGURE 1. Structure of brivaracetam and seletracetam. |
Brivaracetam differs from both levetiracetam and seletracetam in that it appears to possess a slightly broader activity spectrum in animal seizure models, and it is active in the
MES and PTZ seizure tests. These effects of brivaracetam are thought to be related to its modest inhibition of sodium (Na) channels.129,175 Brivaracetam also appears to be more effective than levetiracetam in its ability to suppress both the behavioral seizure and electrographic afterdischarge in the amygdala-kindled rat model of partial epilepsy. In addition to its more potent action against the fully expressed kindled seizure, brivaracetam also appears to be more potent and more effective than levetiracetam in preventing the acquisition of corneal kindling.103
MES and PTZ seizure tests. These effects of brivaracetam are thought to be related to its modest inhibition of sodium (Na) channels.129,175 Brivaracetam also appears to be more effective than levetiracetam in its ability to suppress both the behavioral seizure and electrographic afterdischarge in the amygdala-kindled rat model of partial epilepsy. In addition to its more potent action against the fully expressed kindled seizure, brivaracetam also appears to be more potent and more effective than levetiracetam in preventing the acquisition of corneal kindling.103
At the cellular level, brivaracetam does not directly display any direct effects on γ-aminobutyric acid (GABA)-, glycine-, kainic acid-, or α-amino-3-hydroxy 5 methyl isoxazole 4 propionate (AMPA)-gated currents.128 It has been found to exert a small inhibitory effect on the amplitude of the plateau phase of N-methyl-D-aspartate (NMDA)-evoked currents.127 Seletracetam also appears to be a potent inhibitor of high-voltage activated (HVA) calcium (Ca2+) currents.121 Results from combination studies conducted with seletracetam, ω-conotoxin GVIA and ω-conotoxin MVIIC suggest that this effect of seletracetam is mediated by both N- and Q-type HVA channels. In addition, seletracetam has also been found to reverse the inhibitory effect of zinc and β-carboline inhibition of glycine-gated currents.127 In contrast to brivaracetam, seletracetam is inactive at voltage-gated Na+ channels.175
Low concentrations of brivaracetam (1–10 μM) suppress high potassium (K), low calcium (Ca)-induced epileptiform bursting in hippocampal slices.100 Levetiracetam has been shown to exert a similar action but requires substantially higher (i.e., 32 μM) concentrations. Brivaracetam (3.2 μM) differs from levetiracetam in that it is also capable of reducing spontaneous bursting; whereas levetiracetam is inactive at concentrations up to 32 μM. The ability of brivaracetam to inhibit spontaneous bursting may be related to its inhibitory effect on voltage-activated Na+ currents.176
Pharmacokinetics
To date, published clinical information is available only for brivaracetam. After single doses up to 1,400 mg and multiple doses up to 800 mg/day in healthy volunteers, brivaracetam is rapidly absorbed from the gastrointestinal tract, with a median time of peak concentration at 1 hour.99 Plasma drug levels increase proportionally with dose.131 Concomitant intake of food does not affect extent of exposure, but the rate of absorption is lower and peak concentrations are reduced by about 30%.
The half-life of brivaracetam in healthy volunteers is in the range of 6 to 11 hours (average 7.7 hours).99 Only a minor fraction of the apparent oral clearance (CL/F) of brivaracetam is accounted for by renal clearance of unchanged drug.131 Identified urinary metabolites include 2-(2-oxo-4-propylpyrrolidin-1yl)butyric acid and 2-(2-oxo-4-propylpyr-rolidin-1yl)-4-hydroxybutanamide.99
Efficacy and Adverse Effects
In a Phase II study in 19 photosensitive epilepsy patients, single oral doses of brivaracetam ranging from 10 to 80 mg were effective in reducing the photoparoxysmal electroencephalographic response, which was abolished in 78% of patients.63a,99 The 80 mg dose was most effective in terms of time to reach the maximal response and duration of response. Median time to maximal response for all doses was 0.5 hours and median duration of effect was about 28 hours after 10, 20, or 40 mg, and 59.5 hours after 80 mg.
In healthy volunteers given single doses up to 800 mg/day and multiple doses up to 800 mg/day, adverse events were usually dose-related and included dizziness, somnolence, headache, nausea, and euphoric mood.99 In the photosensitivity study, the adverse effect profile was similar to that reported in healthy volunteers.
Eslicarbazepine Acetate (S-Licarbazepine Acetate, BIA 2-093)
Structure and Chemistry
Eslicarbazepine acetate (S-licarbazepine acetate, BIA 2-093), S-(-)-10-acetoxy-10,11-dihydro-5H-dibenzo/b,f/azepine-5-car-boxamide, is structurally related to carbamazepine and oxcarbazepine in that it shares the dibenzazepine nucleus53 (Fig. 2). In this respect, eslicarbazepine acetate was designed by BIAL, Portugal, to be a potentially improved alternative to carbamazepine and oxcarbazepine. However, as shown in FIGURE 2, it is important to note that oxcarbazepine is metabolized to (S)-licarbazepine (eslicarbazepine) and (R)-licarbazepine in a 4:1 ratio. Thus, the only difference between oxcarbazepine and eslicarbazine acetate is that eslicarbazepine acetate is only metabolized to the (S)-enantiomer with minor chiral conversion to the (R)-enantiomer, whereas oxcarbazepine is converted to both the (S)- and (R)-enantiomers.a Given that there is no evidence to suggest that the (R)-enantiomer is more toxic than the (S)-enantiomer or that it interferes with the efficacy of the (S)-enantiomer, it is not likely that eslicarbazepine acetate will provide any major advantage over oxcarbazepine. In fact, in the MES test, the less potent (R)-enantiomer displays an equivalent, if not more favorable protective index (i.e., the ratio of the median toxic dose in the rotorod test to the median MES efficacious dose) than the (S)-enantiomer.15
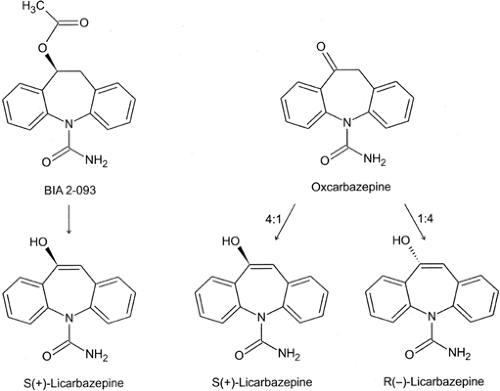 FIGURE 2. Structure of eslicarbazepine acetate (BIA 2-093) and ox-carbazepine. Eslicarbazepine is rap-idly metabolized to its active meta bolite S(+)-licarbazepine. Oxcarbazepine is also converted to its mono-hydroxy derivatives S(+)-licar-bazepine and R(–)-licarbazepine in a 4:1 ratio. (Modified from Rogawski MA. Diverse mechanisms of antiep-ileptic drugs in the development pipeline. Epilepsy Res. 2006;69(3):273–294, with permission.) |
Pharmacology
Eslicarbazepine acetate behaves very much like carbamazepine and oxcarbazepine in animal seizure and epilepsy models.
It is effective against tonic extension seizures induced by MES and focal seizures in the amygdala-kindled rat model of partial epilepsy.15 Like carbamazepine and oxcarbazepine, eslicarbazepine is mostly inactive against clonic seizures induced by PTZ. This activity profile would predict efficacy in humans against generalized tonic–clonic seizures and partial seizures, respectively.168 In these models, eslicarbazepine acetate is equipotent to carbamazepine and more potent than oxcarbazepine.
It is effective against tonic extension seizures induced by MES and focal seizures in the amygdala-kindled rat model of partial epilepsy.15 Like carbamazepine and oxcarbazepine, eslicarbazepine is mostly inactive against clonic seizures induced by PTZ. This activity profile would predict efficacy in humans against generalized tonic–clonic seizures and partial seizures, respectively.168 In these models, eslicarbazepine acetate is equipotent to carbamazepine and more potent than oxcarbazepine.
Mechanistically, eslicarbazepine displays a voltage-depen-dent block of voltage-dependent Na currents.21 In a manner similar to other voltage-sensitive Na channel blockers, eslicarbazepine inhibits Na-dependent neurotransmitter release at concentrations similar to those observed for carba-mazepine and oxcarbazepine.6,116
Pharmacokinetics
After oral administration of single and multiple doses, unchanged eslicarbazepine acetate is generally undetectable (<10 ng/mL) in plasma.4,5 Eslicarbazepine acetate is rapidly and extensively converted to (S)-licarbazepine, whose chemical structure corresponds to the S-enantiomer of 10-hydroxy-carbazepine, the active mono-hydroxy-metabolite of oxcarbazepine.4,5,15,53 Chiral conversion of (S)-licarbazepine to (R)-licarbazepine is minor, and only about 5% of circulating licarbazepine is in the R-form2,18; by contrast, after administration of oxcarbazepine, the ratio between (R)- and (S)-licarbazepine in blood is on the order of 20% to 25%.164
Peak plasma concentrations of licarbazepine occur 0.75 to 4 hours after single oral doses of eslicarbazepine acetate ranging between 20 and 1,200 mg.4 The bioavailability of licarbazepine, measured in terms of area under the curve (AUC), is about 16% greater than that observed after an equivalent molar dose of oxcarbazepine.18 The pharmacokinetic profile of licarbazepine is not affected by coadministration of eslicarbazepine acetate with food.98 Bioequivalence between a suspension and a tablet form has been demonstrated.49
During multiple doses ranging from 200 mg twice a day to 2,400 mg once daily, peak licarbazepine concentrations are usually observed 2 to 3 hours after each dose intake, and steady-state concentrations are reached within 4 to 5 days of once or twice daily administration.5,162 The extent of systemic exposure to licarbazepine increases in an approximately dose-proportional manner after both single and multiple doses.
The half-life of licarbazepine has been estimated to be in the range of 8 to 17 hours after single doses and 9 to 13 hours after multiple dosing,2,4,5 although an “effective” half-life of 17 to 24 hours has been inferred from the time taken to reach steady-state.2,5 Licarbazepine is excreted in urine in free and conjugated form, with about 20% and 40% of the dose being recovered within 12 and 24 hours post-dose respectively.3,4,5 Oxcarbazepine has been detected as a minor metabolite.4
In a study that compared the plasma levels of (R)- and (S)-licarbazepine after single (600 mg) and multiple doses (600 mg once daily for 8 days) in 12 young (mean age 30 years, range 18–38) and 12 elderly (mean age 70 years, range 65–80) healthy volunteers, no pharmacokinetic differences were identified between the two age groups.2
Drug Interactions
Because licarbazepine clearance in patients taking oxcarbazepine is known to be moderately increased by coadministration of enzyme-inducing AEDs,106 it is reasonable to assume that a similar interaction may apply to licarbazepine derived from eslicarbazepine acetate.
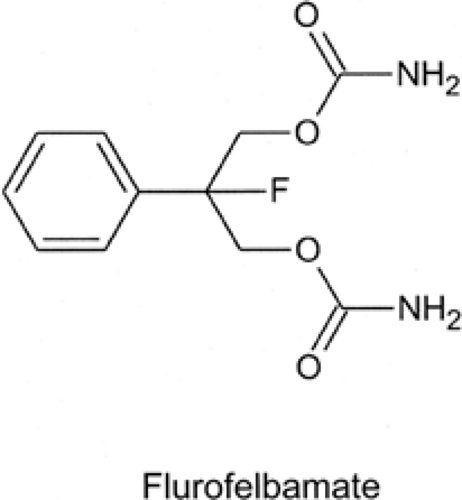 FIGURE 3. Structure of fluorofelbamate. |
In healthy subjects, eslicarbazepine acetate at a dosage of 1,200 mg/day did not affect the pharmacokinetics of digoxin to a clinically significant extent.97 Whether eslicarbazepine acetate shares with oxcarbazepine the ability to stimulate the metabolism of steroid oral contraceptives and other substrates has not been reported.
Clinical Efficacy
A multicenter double-blind, parallel-group, adjunctive-therapy study compared once daily and twice daily eslicarbazepine acetate (at escalating doses of 400, 800, and 1,200 mg/day, each for 4-week periods) with placebo in 143 patients with refractory partial seizures.18,96 The proportion of responders (defined as patients showing at least 50% seizure reduction) during the 800- and 1,200-mg/day periods were significantly higher in the once-daily group than in the placebo group, and there were no statistically significant differences in responder rates between the once- and twice-daily group.
Adverse Effects
In healthy volunteers, single and multiple doses of eslicarbazepine acetate up to 1,200 mg and 1,200 mg/day, respectively were tolerated well.3,5 In the adjunctive therapy trial, tolerability was rated as good or very good in 90% of patients in the once-daily group, 80% in the twice-daily group, and 83% in the placebo group.18
Fluorofelbamate
Structure and Chemistry
Fluorofelbamate (2-phenyl-2-fluoro-1,3 propanediol dicarbamate) is a felbamate analog designed to have the clinical efficacy of felbamate without its serious adverse effects (i.e., aplastic anemia and hepatic toxicity). Structurally, fluorofelbamate differs from felbamate in that it possesses a fluorine atom instead of a hydrogen at the 2-position of the propanediol moiety (Fig. 3). This substitution is thought to result in a more stable molecule and to prevent the production of atropaldehyde (ATPAL, or 2-phenylpropenal), the proposed toxic reactive metabolite of felbamate.154 Fluorofelbamate is being developed by MedPointe Healthcare, Inc.
Pharmacology
Fluorofelbamate, like felbamate, is active against a variety of electrically and chemically induced seizures in animal models.18 In addition to blocking tonic extension in the MES test, it also blocks clonic seizures induced by picrotoxin, and sound-induced seizures in the Frings mouse. Fluorofelbamate (100 and 200 mg/kg) is also quite effective in reducing the cumulative seizure duration in the perforant path model of self-sustaining status epilepticus (SE) when administered early. Slightly higher doses (i.e., 200 and 300 mg/kg) attenuated seizures in the self-sustaining status model when administered late and at a time when the status was refractory to treatment with conventional anticonvulsants.107
In addition to is anticonvulsant activity, fluorofelbamate has been reported to be neuroprotective in several models. For example, it decreases neuronal damage in an in vitro chemical model of ischemia (i.e., NaCN), protects against hypoxic CA1 hippocampal neuronal damage in an in vitro slice model, decreases the infarct volume in an in vivo rat pup model of hypoxia, and reduces CA1 damage associated with transient global ischemia in gerbils.18,166
Limited in vitro data are available regarding the mechanism of action of fluorofelbamate. Based on the electrophysiologic studies thus far reported, it would appear that fluorofelbamate is not a potent modulator of either inhibitory- or excitatory-mediated neurotransmission. It has been reported to produce a modest inhibition of kainate- and NMDA-mediated currents in mouse cortical neurons and to exert a modest inhibition of voltage-dependent Na currents in NIE-115 neuroblastoma cells.18,129 It is not clear whether these modest effects are sufficient to account for its seemingly broad anticonvulsant and neuroprotective profile in animal models.
Pharmacokinetics
Based on evidence suggesting that a reactive metabolite, ATPAL, may be responsible for the toxicities observed during therapy with felbamate, Parker et al.117 tested the hypothesis that fluorofelbamate would not be metabolized to ATPAL. They compared the metabolism by in vitro human liver postmitochondrial suspensions (S9) of selected felbamate and postulated fluorofelbamate metabolites and concluded that fluorofelbamate is not metabolized in vitro by S9 to ATPAL.
In pharmacokinetic studies in rats and dogs, exposure increased proportionally with dose. In dogs, peak plasma concentrations were reached in 2 to 6 hours, and the terminal half-life was estimated at 6 to 9 hours.18 No studies in humans have been reported to establish fluorofelbamate pharmacokinetics, interaction potential, efficacy, or adverse-effect profiles.
Ganaxolone (CCD 1042)
Structure and Chemistry
Ganaxolone, 3α-hydroxy-3β-methyl-5 α-pregnan-20-one, is the 3β-methyl analog of the endogenous steroid allopregnanolone and is a potent positive modulator of GABAA receptors. It is currently under development by Marinus Pharmaceuticals (Fig. 4).
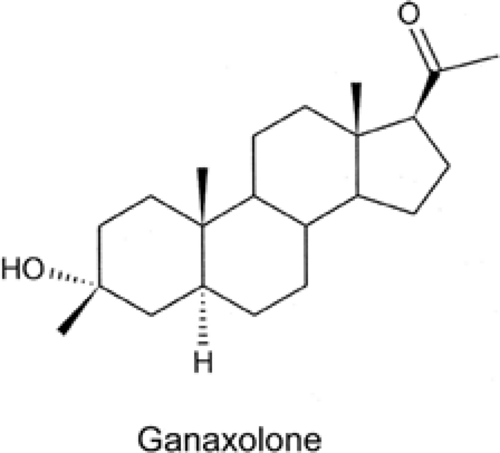 FIGURE 4. Structure of ganaxolone. |
Pharmacology
In seizure and epilepsy models, ganaxolone is effective against clonic seizures induced by PTZ and bicuculline.25 At nontoxic
doses, ganaxolone is less active against tonic extension seizures induced by MES. It is also active against seizures induced by aminophylline, τ-butyl-bicyclo-phosphorothionate, and fluorothyl (see Perucca and Kupferberg120a, for review and references). Ganaxolone has also shown activity in the 6-Hz model of pharmacoresistent partial epilepsy,61 is active in acute SE models,70 and demonstrated efficacy against partial seizures in the corneal- and amygdala-kindled rat models.25,126 In contrast to the benzodiazepines, the anticonvulsant activity of ganaxolone in animal models is not diminished with chronic treatment.
doses, ganaxolone is less active against tonic extension seizures induced by MES. It is also active against seizures induced by aminophylline, τ-butyl-bicyclo-phosphorothionate, and fluorothyl (see Perucca and Kupferberg120a, for review and references). Ganaxolone has also shown activity in the 6-Hz model of pharmacoresistent partial epilepsy,61 is active in acute SE models,70 and demonstrated efficacy against partial seizures in the corneal- and amygdala-kindled rat models.25,126 In contrast to the benzodiazepines, the anticonvulsant activity of ganaxolone in animal models is not diminished with chronic treatment.
Ganaxolone is a potent positive modulator of GABAA-evoked currents. It is active at GABAA receptors containing most αsubunits, including α1, α2, α3, α4, and α6. Unlike the benzodiazepines, the activity of most neurosteroids at the GABAA receptor is not dependent on the presence of a γ2 subunit.25 Interestingly, GABA receptors containing a δ-subunit are particularly sensitive to modulation by neurosteroids.129,169 Because of their presynaptic and extrasynaptic localization,43,113 δ-subunits containing GABAA receptors are ideally suited to mediate tonic currents and may represent an important molecular target for ganaxolone and other potential neurosteroids.129
Pharmacokinetics
Seven different studies in normal volunteers were performed examining the pharmacokinetics of ganaxolone after single (50 to 1,500 mg) and multiple dose regimens (≤300 b.i.d. for 10 days or 500 mg once daily for 2 weeks).110 Because ganaxolone is highly insoluble in aqueous media, it has been formulated as a solution complexed with β-cyclodextrin or 2-hydroxypropyl-β-cyclodextrin to enhance bioavailability.
After oral dosing, ganaxolone is absorbed rapidly, and peak plasma levels are generally attained within 3 hours. Absorption is followed by a biphasic elimination phase, with a rapid decline in plasma concentration over the first 12 hours and a subsequent slower decline characterized by a terminal half-life of 37 to 70 hours.110 Plasma concentrations after multiple doses are similar in shape to those observed after single doses, with a sharp decrease in plasma levels over the first 12 hours. Accumulation is limited, as indicated by a lack of increasing trough levels during repeated dosing. The pharmacokinetics of ganaxolone appear to be linear, with mean peak plasma levels at steady state ranging from 32 ng/mL at 50 mg once daily to 376 ng/mL at 500 mg once daily.
Drug Interactions
In children aged 7 months to 7 years with infantile spasms, no consistent changes in the serum levels of concomitant AEDs with administration of ganaxolone were observed.66
Clinical Efficacy
A double-blind, randomized, placebo-controlled trial to examine the safety, tolerability, and antiepileptic activity of ganaxolone after withdrawal from other AEDs during presurgical evaluations was performed in 52 patients who were randomized to receive ganaxolone (24 patients) or placebo (28 patients) for up to 8 days.78 Ganaxolone was administered at a dose of 1,500 mg/day on day 1 and 1,875 mg/day on days 2 to 8. Dosing occurred three times per day. The primary endpoint was time to withdrawal from the trial. Intent-to-treat survival analyses revealed a trend toward efficacy with ganaxolone (p = 0.0795, log rank test). Kaplan-Meier curves depicted a clear separation between treatment groups, with 50% of the ganaxolone-treated patients completing the entire study, compared with 25% of patients treated with placebo. Covariate analyses revealed a significant treatment effect on survival time in men (p = 0.03). Post-hoc analyses focusing on patients who completed the entire study revealed a significant difference (p = 0.04) between treatment groups. A clear relationship between trough serum concentrations and efficacy was not evident.
A multicenter, open-label, add-on trial investigated the safety and efficacy of ganaxolone in 20 children (age 7 months to 7 years) with refractory infantile spasms, or with continuous seizures after a prior history of infantile spasms.66 The dose of ganaxolone was increased to 36 mg/kg per day (or to the maximally tolerated dose) over 4 weeks, then maintained for 8 weeks before tapering off. The frequency of spasms was reduced by at least 50% in 33% of the subjects with spasms, with an additional 33% experiencing some improvement (25%–50% reduction in spasms frequency). In these subjects with highly variable sampling times, a ganaxolone concentration-response curve could not be established.
Adverse Effects
In normal volunteers, ganaxolone was well tolerated after single doses (≤1,500 mg) and multiple doses (≤300 mg b.i.d. for 10 days).110 No serious or life-threatening adverse events attributed to the drug were observed. The majority of adverse events were mild (82%) to moderate (14%) and were limited to headache, dizziness, somnolence, gastrointestinal disturbances, and malaise. There was no apparent effect on electrocardiogram (ECG) or vital signs.
In the presurgical study of Laxer et al.,78 47 adverse events were reported in the ganaxolone group and 38 in the placebo group. Central nervous system (CNS) adverse effects occurred in both groups, with dizziness being the most common complaint. Two serious adverse events occurred; one patient receiving ganaxolone experienced agitation and depression on the first day of treatment, and one patient receiving placebo experienced postictal psychosis.
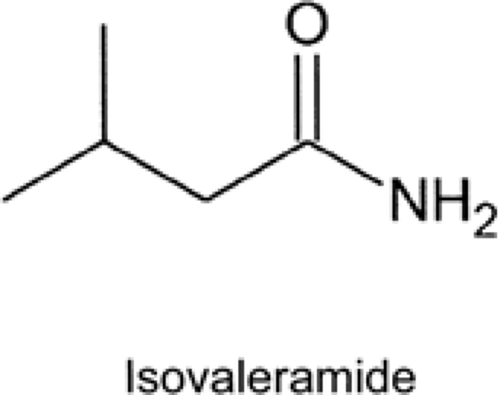 FIGURE 5. Structure of isovaleramide. |
In the infantile spasms study, the most common adverse events thought to be possibly, probably, or definitely related to ganaxolone were somnolence, diarrhea, nervousness, and vomiting.66 In another open-label pediatric study in 15 children aged 5 to 15 years treated with doses up to 12 mg/kg three times a day, adverse events included somnolence, sleep disturbances,
nervousness, constipation, change in seizure types and, in one case, disturbed behavior and cognition.79
nervousness, constipation, change in seizure types and, in one case, disturbed behavior and cognition.79
Isovaleramide (NPS 1776)
Structure and Chemistry
Isovaleramide (NPS 1776), 3-methylbutanamide, is a low-molecular-weight, branched-chain amide that displays a broad-spectrum of activity in animal models (Fig. 5). It is being developed by NPS Pharmaceuticals.
Pharmacology
In animal seizure and epilepsy models, NPS 1776 possesses an anticonvulsant profile similar to that of valproate (VPA). It is orally active against tonic extension seizures in the MES test and clonic seizures induced by PTZ, bicuculline, and picrotoxin. Furthermore, it is effective in reducing the behavioral seizure score in both the corneal- and amygdala-kindled rat models of partial epilepsy, and it reduces the afterdischarge duration in the amygdala-kindled rat. Daily dosing with NPS 1776 has also been found to delay the acquisition of kindling in the amygdala-kindled rat, thereby suggesting that this compound, like VPA, may exhibit antiepileptogenic effects. NPS 1776 is also effective in two genetic seizure models (i.e., the Frings audiogenic seizure susceptible mouse and the Wistar rat). In the Wistar rat, NPS 1776 was found to reduce the spontaneous electrographic spike-and-wave discharges. In a chronic dosing study in the Frings audiogenic seizure-susceptible mouse, no sign of tolerance was observed following daily dosing with NPS 1776 for 28 days. Overall, the preclinical anticonvulsant profile of NPS 1776 suggests that it will have broad-spectrum efficacy against partial and generalized seizures.16,18,167 In addition to its anticonvulsant action, NPS 1776 has been found to possess antispastic, analgesic, and anxiolytic activity.18
To date, the mechanism of action of NPS 1776 has not been elucidated. It is inactive in in vitro binding and uptake assays against a number of receptor- and voltage-active ion channels at a concentration of 1,000 μM, suggesting that it does not exert an action through a direct interaction at the sites studied.18
 FIGURE 6. Structure of lacosamide. |
Pharmacokinetics
After single and multiple doses up to 1,600 mg and 2,400 mg/day, respectively in healthy volunteers, isovaleramide is rapidly absorbed from the gastrointestinal tract.18 The increase in systemic exposure with increasing dose is slightly more than dose-proportional after single doses, but no major deviations from linearity were observed after multiple dosing.
Isovaleramide is negligibly bound to plasma proteins and is rapidly eliminated with a half-life of 2 to 4 hours. Less than 4% of an orally administered dose is excreted unchanged in urine, and elimination is primarily metabolic. A sustained-release formulation is under development.18
Efficacy and Tolerability
Isovaleramide has shown excellent tolerability in Phase I studies in healthy volunteers. Clinical studies have been initiated to assess its potential value in the treatment of epilepsy and in the acute treatment of migraine, but no results have been reported to date.18
Lacosamide (Harkoseride, SPM 927)
Structure and Chemistry
Lacosamide, also formerly known as harkoseride and SPM927, is the R-enantiomer of 2-acetamido-N-benzyl-3-methoxy-propionamide. It is currently under development by Schwarz BioSciences for the treatment of partial epilepsy and neuropathic pain (Fig. 6).
Pharmacology
Lacosamide displays broad-spectrum anticonvulsant activity in rodent models of generalized and partial epilepsy.18,148 It is effective against audiogenic seizures in Frings mice and is a potent blocker of MES-induced tonic extension seizures in mice and rats. Lacosamide is less effective against clonic seizures induced by PTZ, but it is quite potent against limbic seizures in the 6-Hz psychomotor seizure model of pharmacoresistant epilepsy.40 Lacosamide is also very effective in blocking both the behavioral seizure and afterdischarge duration in the hippocampal-kindled rat model of partial epilepsy. Last, lacosamide is effective in reducing the cumulative seizure duration in the perforant path model of self-sustaining SE. In addition to its effects in seizure and epilepsy models, lacosamide has demonstrated
an ability to attenuate the acquisition of amygdala kindling and displays in vitro and in vivo neuroprotective properties. For example, lacosamide has been shown to delay the development of amygdala kindling at doses of 10 and 30 mg/kg.23 Furthermore, it decreased the infarct volume in the rat middle cerebral artery occlusion (MCAO) model of ischemia when administered 15 minutes before MCAO and for 4 hours postinfusion.122 In contrast, lacosamide was ineffective in preventing brain damage or functional deficits in a rat model of traumatic brain injury.122 In organotypic rat hippocampal slice cultures, lacosamide produced a concentration-dependent block of glutamate- and oxygen-glucose deprivation–induced apoptosis. These latter effects might suggest that lacosamide possesses a disease-modifying effect; however, additional studies are required to fully evaluate this potential attribute.
an ability to attenuate the acquisition of amygdala kindling and displays in vitro and in vivo neuroprotective properties. For example, lacosamide has been shown to delay the development of amygdala kindling at doses of 10 and 30 mg/kg.23 Furthermore, it decreased the infarct volume in the rat middle cerebral artery occlusion (MCAO) model of ischemia when administered 15 minutes before MCAO and for 4 hours postinfusion.122 In contrast, lacosamide was ineffective in preventing brain damage or functional deficits in a rat model of traumatic brain injury.122 In organotypic rat hippocampal slice cultures, lacosamide produced a concentration-dependent block of glutamate- and oxygen-glucose deprivation–induced apoptosis. These latter effects might suggest that lacosamide possesses a disease-modifying effect; however, additional studies are required to fully evaluate this potential attribute.
Lacosamide is effective in various animal models of chronic pain.149 For example, it is active in the formalin, carrageenan model, and the adjuvant-induced arthritis models of chronic pain. The results from these studies support the ongoing clinical investigations in the treatment of pain associated with diabetic neuropathy.
From a mechanistic perspective, more can be said about what lacosamide does not do at the cellular level than what it does do. To date, little is known about the molecular mechanism underlying its broad-spectrum anticonvulsant profile.129 Lacosamide does not directly interact with GABAergic or glutamatergic neurotransmission. It does not block sustained repetitive firing or voltage-dependent Na currents. Furthermore, it does not enhance K currents or attenuate Ca currents. Lacosamide does appear to block slow but not fast depolarization-induced action potential firing in cultured cortical neurons. Like carbamazepine, lacosamide is effective in the 4-amino-pyridine (4-AP) model of epileptiform bursting, and its activity in this model, unlike that of carbamazepine, is completely reversible.80 The results obtained to date suggest that lacosamide has a potentially unique mechanism of action relative to a majority of the available AEDs.
Pharmacokinetics
Lacosamide is efficiently absorbed from the gastrointestinal tract, with peak plasma concentrations being achieved within 1 to 5 hours of dosing.18,75 Oral bioavailability is virtually complete, as documented by the observation that plasma lacosamide levels after the administration of the same dose using either oral tablets or 30- or 60-minute intravenous infusions meet criteria for bioequivalence.75,132 Concomitant intake of food has no effect on the rate or extent of lacosamide absorption,27 and plasma concentrations are linearly related to dose over the 100- to 800-mg dose range.132
Lacosamide is negligibly (<10%) bound to plasma proteins.18 The half-life is on the order of 12 to 16 hours, and elimination occurs partly by renal excretion in unchanged form and partly by biotransformation. About 40% of an orally administered dose is recovered unchanged in urine, and another 30% is recovered in the form of the inactive O-dimethyl-metabolite SPM 12809.18,75,140 In a study designed to assess the potential role of CYP2C19 on lacosamide disposition, plasma lacosamide levels after oral and intravenous administration were similar in eight extensive metabolizers and three poor metabolizers for CYP2C19. However, the plasma levels of the O-demethyl-metabolite were markedly reduced in the poor metabolizers, suggesting that CYP2C19 contributes to lacosamide demethylation.74
After normalization for dose and differences in body weight, plasma lacosamide concentrations do not differ significantly between males and females. In a multiple-dose study, plasma lacosamide concentrations at steady-state in 23 elderly subjects (≥65 years, age range not stated) were about 10% to 35% higher than those observed in a similar cohort of subjects aged 18 to 45 years.140
Drug Interactions
In studies conducted in healthy volunteers, lacosamide pharmacokinetics were not affected by coadministration of carbamazepine 400 mg/day or valproic acid 600 mg/day.74
In vitro, lacosamide shows no potential to inhibit CYP1A2, CYP2A6, CYP2C9, CYP2D6, CY2E1, and CYP3A4 in human hepatocytes.74 In studies with human recombinant CYPs, lacosamide inhibits CYP2C19, but the median inhibitory concentration (0.45 mg/mL) is greater than the anticipated human therapeutic plasma levels. Lacosamide does not induce CYP1A2 and CYP3A4 activity in human hepatocytes.
In healthy volunteers, lacosamide (400 mg/day) has been found not to affect the pharmacokinetics of carbamazepine, valproic acid, metformin, digoxin, ethinylestradiol, and levo-norgestrel. Likewise, clinical trials in patients receiving adjunctive treatment with lacosamide at dosages up to 600 mg/day did not identify any change in serum levels of carbamazepine, carbamazepine-10,11-epoxide, phenytoin, valproic acid, lamotrigine, mono-hydroxy-carbazepine, topiramate, zonisamide, levetiracetam, and gabapentin.18,59,74
Clinical Efficacy
Studies in Epilepsy
A multicenter, double-blind, randomized, parallel-group, placebo-controlled trial assessed lacosamide (200, 400, and 600 mg/day in two divided doses) as adjunctive therapy in 418 patients with refractory partial-onset seizures.14,59 Lacosamide was titrated to target dose in 100 mg/day increments at 1-week intervals, followed by a 12-week maintenance phase. In the full analysis (ITT) data set, median reduction in seizure frequency (maintenance vs. baseline) was 10% on placebo, 26% at 200 mg/day, 39% at 400 mg/day, and 40% at 600 mg/day. Responder rates (proportion of patients with at least 50% seizure reduction) was 22% on placebo and 33%, 41%, and 38% at 200, 400, and 600 mg/day, respectively. Response rates at the 400- and 600-mg dose were statistically superior to placebo. The proportion of patients completing the maintenance period without major protocol deviations was 78%, 74%, and 60% in the 200, 400, and 600 mg/day groups, respectively, compared with 88% in the placebo group.59 Follow-up observations in patients who continued open-label lacosamide after completion of the double-blind phase suggest that efficacy is maintained during long-term treatment.35
Studies in Neuropathic Pain
Lacosamide has been investigated in two multicenter, double-blind, randomized, parallel-group, placebo-controlled trials in patients with neuropathic pain attributed to distal diabetic neuropathy. One trial explored dosages of 200, 400, and 600 mg/day in a total of 370 patients,170 while the second trial assessed 400 and 600 mg/day in 357 patients.174 The study design involved gradual titration to the target dose and a 12-week maintenance period. In both studies, the best response was in the 400 mg/day dose groups. In one of the trials, the reduction in pain score in the 400-mg group was statistically significant compared with placebo,170 while in the other the difference compared with placebo during the last 4 weeks of treatment failed to reach statistical significance.174 Administration of a
600 mg/day dose did not appear to confer additional benefit in reducing pain scores, and was less tolerated.
600 mg/day dose did not appear to confer additional benefit in reducing pain scores, and was less tolerated.
Adverse Effects
In the adjunctive-therapy placebo-controlled trial in patients with partial onset seizures, the most commonly observed adverse events were dizziness, headache, fatigue, ataxia, gastrointestinal disturbances (nausea, vomiting), and visual disturbances (blurred vision, diplopia, and nystagmus), most of which were dose-related.14 The most common of all events was dizziness, which was reported by 24%, 26%, and 55% of patients at 200, 400, and 600 mg/day, respectively, compared with 10% on placebo. The proportion of patients withdrawing due to adverse events was 5% on placebo, 11% at 200 mg/day, 19% at 400 mg/day, and 30% at 600 mg/day. Remarkably, there were little differences in proportion of patients reporting somnolence between the placebo and any lacosamide dose group.
A formulation of lacosamide for intravenous use is being developed to allow replacement therapy for patients temporarily unable to continue oral therapy. In healthy volunteers, adverse events after infusion of 200 mg lacosamide over periods of 15, 30, and 60 minutes were similar to those observed after intake of a 200-mg tablet.75 Likewise, adverse events associated with 30- to 60-minute infusions (up to 300 mg b.i.d. for two days) given as replacement treatment in patients with epilepsy stabilized on oral lacosamide were comparable to those observed after oral dosing.18,132
Studies in Neuropathic Pain
In the first trial conducted in patients with neuropathic pain, the proportion of patients discontinuing treatment due to adverse events was 8.6%, 23.1%, and 39.8% in the placebo, 200, 400, and 600 mg/day groups, respectively.170 In the second trial, discontinuation rates were 11.3% and 23.3% at 400 and 600 mg/day, respectively, compared with 5.4% on placebo.174 In both trials, lacosamide was generally well tolerated, with similar rates of adverse events in the placebo and active treatment groups at doses up to 400 mg/day. In the 600 mg/day group, the incidence of dizziness (29.0% in the first study and 19.5% in the second study) was considerably higher than in the other groups.
Losigamone
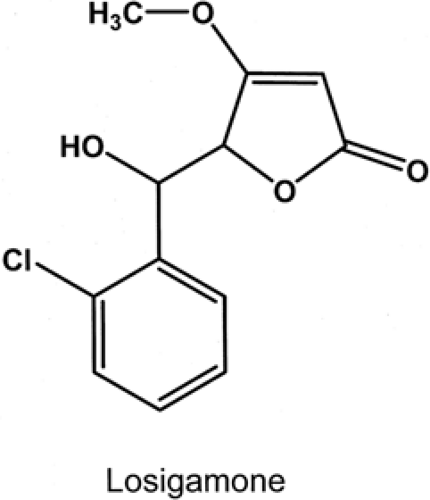 FIGURE 7. Structure of losigamone. |
Structure and Chemistry
Losigamone, threo ±5(S,R)-5-[(2-chlorophenyl)hydroxyl-methyl)]-4-methoxy(5H)-furanone, is a racemic mixture that was synthesized at the Willmar Schwabe Company in Karlsruhe, Germany (Fig. 7).
Pharmacology
In most pharmacologic studies conducted to date, the S(+)-enantiomer of losigamone is more potent than the R(-)-enantiomer.173 In animals, losigamone is active in blocking tonic hind limb extension induced by a number of stimuli including MES, PTZ, bicuculline, nicotine, and 4-aminopyridine but not strychnine or picrotoxin.146 It is also effective against clonic seizures induced by PTZ, bicuculline, and picrotoxin.146 In addition, losigamone has been shown to inhibit audiogenic seizures in rats and gerbils and to attenuate behavioral seizures in the PTZ-kindled mouse.120
Like many of the AEDs, the precise mechanism of action of losigamone has yet to be established. In the in vitro hippocampal slice preparation, losigamone has been demonstrated to reduce the frequency and amplitude of picrotoxin-, low Ca2+/low magnesium (Mg2+), and low Ca2+/low Mg2+/high K+-induced seizure-like events in a concentration-dependent manner.68,69,81,146 At higher concentrations, losigamone has been found to decrease the late recurrent epileptiform discharges in area CA1 and CA3 of the hippocampus and the ictiform events in the entorhinal cortex that develop following prolonged exposure to low-Mg2+ containing buffer.173 Losigamone has also been shown to decrease depolarization-induced repetitive spike firing and stimulus-induced excitatory postsynaptic potentials in the entorhinal cortex slice preparation141 and 4-aminopyridine-induced epileptiform activity in rat hippocampal slice preparation.172 Losigamone does not appear to modify either fast or slow inhibitory postsynaptic potentials.141
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


