Ketogenic Diet
Carl E. Stafstrom
Eileen P. G. Vining
Jong M. Rho
Introduction
The ketogenic diet (KD) is a high-fat, low-carbohydrate, adequate-protein diet that has been used as an alternative therapy for intractable seizures since the early 1920s. At that time, only a few effective antiepileptic drugs (i.e., phenobarbital and bromides) were available to clinicians, and each of these carried significant side effects. In the 1940s and 1950s, when modern antiepileptic drugs such as phenytoin and ethosuximide became available, use of the KD declined rapidly. Nevertheless, there was an occasional resurgence of interest in the KD. In 1971, the medium-chain-triglyceride diet34 was devised in an attempt to provide more flexibility with respect to carbohydrates, enhanced ketosis, and better palatability. Despite a flurry of new antiepileptic drugs in the 1990s, frustration with the medical intractability of childhood epilepsies has continued, and recently, public awareness of the KD has increased. The KD is now considered a safe and effective alternative therapy for children and adults with refractory epilepsy, with an efficacy that rivals (and perhaps exceeds) the newer antiepileptic drugs (AEDs).83 More recently, alternative diets for epilepsy treatment, such as the Atkins diet and a low-glycemic-index diet, have been devised that represent modifications of the original KD.
History
Fasting for seizure control was reported as early as the 5th century BC by Hippocrates.95 References to fasting for seizure control are also found in the Bible (Matthew 17:14-21 and Mark 9:14-29). Jesus, after curing a young boy with convulsions, advised his disciples that such “demons” cannot be purged except by prayer and fasting. Fasting is mentioned again in the medieval writings of John of Gaddesden.50 There is no further mention of fasting in modern medical literature until 1911, when Guelpa and Marie30 placed 20 patients on a 4-day fast, noted some improvement in seizure control, and referred to people with epilepsy as trop alimentés (“too well fed”).
In 1921, Geyelin28 reported that the beneficial effect of 3 weeks of fasting continued even after the fast was broken. The work of Geyelin and many others was probably prompted by reports of a young boy whose seizures were treated successfully with prolonged fasting by an osteopathic practitioner, Hugh Conklin. Conklin was a follower of the physical culturalist Bernard Macfadden.95 Their regimen consisted of 3 to 4 weeks of fasting with only water allowed.49
Based on the observations by Geyelin, Wilder96 theorized that the benefit achieved by fasting might depend on the resultant ketonemia. Utilizing earlier work showing that ketogenesis could be produced by diets very rich in fat and very low in carbohydrate, he proposed a formulation of the KD that is very similar to the one used currently. In 1922, Wilder and Winter97 showed that ketosis occurred when the ratio of fatty acids to glucose in the diet was >2:1, and they recommended that to maintain ketosis, a ketogenic:antiketogenic (K:AK) ratio of at least 3:1 be used. Peterman,67,68 working at the Mayo Clinic, revised the diet and determined minimal daily requirements for calories (75% of the Recommended Daily Allowance for a child’s height and weight) and protein (1 g/kg body weight).
Studies of KD efficacy for seizure control were published in the 1920s by clinicians at the Mayo Clinic. Peterman reported seizure freedom in 50% of patients67; Helmholz and Keith found a third of patients to be seizure free and more than 50% showed “definite improvement” in seizure frequency.32 These successes led several investigators to study the mechanism of action of fasting and the KD in patients using biochemical analyses.8,27,48
Mechanism of Action
Various hypotheses have been put forth over the years to explain KD efficacy, including acidosis, cellular and extracellular dehydration, a direct action of ketone bodies (acetoacetate, β-hydroxybutyrate, acetone) on neuronal firing or synaptic function, effects of lipids on neuronal excitability, changes in the source or utilization of energy within the brain, and alterations of mitochondrial metabolism. None of these is a sufficient explanation for the effectiveness of the KD. The challenge has been to understand how an anticonvulsant effect results from a metabolic shift from carbohydrate to fat as an energy source.21a
There are multiple sites within relevant biochemical pathways where seizure suppression could be facilitated, and the mechanism by which the KD exerts an anticonvulsant effect likely involves the combination of altered energy homeostasis and regulation of neuronal and synaptic excitability. It is also conceivable that the KD may work in different ways against different seizure types and epileptic syndromes. Both clinical studies and animal experiments have been used to decipher KD mechanisms.79 Any explanation of mechanism of action must take into account clinical observations and known biochemical alterations resulting from ingestion of the KD (Table 1).
Overview of Brain Energy Metabolism
Since the original formulation of the KD, it has been assumed that fasting and the KD share a common mechanism in alleviating seizures. The first step in understanding how the KD may work as an antiepileptic treatment is to understand how ketosis occurs. Whether the antiepileptic effectiveness of fasting or the high-fat diet is related to the level of ketosis remains a lingering controversy.
Table 1 Animal Models of the Ketogenic Diet: Observations and Clinical Correlates | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Ordinarily, the brain is an obligate user of glucose as its energy source. However, when carbohydrates are restricted, as in fasting or the KD, the brain can no longer use glucose exclusively. Metabolism shifts such that the brain preferentially oxidizes ketone bodies derived from fats, instead of carbohydrates, as the primary fuel source.17,63,77 Dietary fats are ordinarily
broken down by β-oxidation in the liver into two-carbon acetyl CoA molecules (Fig. 1A), which then enter the tricarboxylic acid (TCA) cycle, producing energy via generation of adenosine triphosphate (ATP). However, under conditions of fasting, starvation, or the high-fat KD, acetyl CoA entry into the TCA cycle is reduced because of low availability of the TCA rate-limiting substrate oxaloacetate and the rate-limiting enzyme α-ketoglutarate dehydrogenase (which have been diverted to gluconeogenesis). Instead, acetyl CoA is used to synthesize the four-carbon ketone bodies, β-hydroxybutyrate (BHB) and acetoacetate (AcAc), as well as the volatile ketone acetone, which is exhaled with fasting, and the KD produces ketosis (i.e., elevated blood levels of BHB and AcAc). The liver lacks the enzymes necessary to break down ketone bodies, so BHB and AcAc are exported in the circulation to tissues such as muscle and brain.
broken down by β-oxidation in the liver into two-carbon acetyl CoA molecules (Fig. 1A), which then enter the tricarboxylic acid (TCA) cycle, producing energy via generation of adenosine triphosphate (ATP). However, under conditions of fasting, starvation, or the high-fat KD, acetyl CoA entry into the TCA cycle is reduced because of low availability of the TCA rate-limiting substrate oxaloacetate and the rate-limiting enzyme α-ketoglutarate dehydrogenase (which have been diverted to gluconeogenesis). Instead, acetyl CoA is used to synthesize the four-carbon ketone bodies, β-hydroxybutyrate (BHB) and acetoacetate (AcAc), as well as the volatile ketone acetone, which is exhaled with fasting, and the KD produces ketosis (i.e., elevated blood levels of BHB and AcAc). The liver lacks the enzymes necessary to break down ketone bodies, so BHB and AcAc are exported in the circulation to tissues such as muscle and brain.
Therefore, during fasting or high-fat/low-carbohydrate intake, the brain can utilize ketones as the main oxidizable substrate for cerebral metabolism (Fig. 1B).18,77 Ketones are transported across the blood–brain barrier by facilitated diffusion, using a monocarboxylate transporter66 induced by fasting. Brain monocarboxylate transporter levels are upregulated by KD treatment.14,47
Neurons and glia possess the requisite enzymes to break down BHB and AcAc into acetyl CoA fragments (BHB dehydrogenase and 3-oxoacid-CoA transferase, respectively). Acetyl CoA molecules can in turn enter the TCA cycle and produce energy (Fig. 1B). These enzymes are regulated developmentally, with maximal expression early in life, consistent with the better utilization of ketones at young ages.14 Although the brain can metabolize ketones if deprived of its usual carbohydrate fuels, cerebral activity may be optimal only if some glucose is present. A recent study showed that cortex, brainstem, and primary cultures of cortical astrocytes can express ketogenic enzymes, especially early in development.13
Ketones
A reasonable hypothesis is that ketone bodies exert anticonvulsant activity, much like antiepileptic drugs. Serum ketone concentrations rise markedly in subjects on the diet and urinary ketones are used clinically to indicate ketosis. However, several clinical and experimental studies have shown that the relationship between serum or urinary ketones and seizure control is imprecise.7,29,33,53 Direct effects of ketones on excitatory and/or inhibitory neurotransmission and on ictal activity in vitro have been disappointingly negative.78,87 However, in vivo, rats on the KD or a calorie-restricted normal diet exhibited increased paired-pulse inhibition in the hippocampal dentate gyrus and increased resistance to maximal dentate activation (a form of seizure activity).6 Acetone could also exert an anticonvulsant effect.51,52,54,59,60,72 In summary, there appears to be a “threshold level” of ketosis that must be achieved and maintained for the KD to be maximally effective.
Alterations in Energy Metabolism and Mitochondrial Function
The alteration in cerebral energetics induced by the KD favors an increase in “energy charge” (i.e., a relative increase in the ATP:ADP ratio resulting from metabolic alterations of the enzymes involved in glycolysis and the TCA cycle17). The greater availability of brain energy may reduce cellular excitability by enhancing energy available for cellular processes such as membrane pumps and transporters, which enhance hyperpolarization. The “energy charge” hypothesis is supported by recent studies in fasting humans using 31P spectroscopic imaging, in which ketones are transported from the blood to the brain and are utilized by neurons.64,65
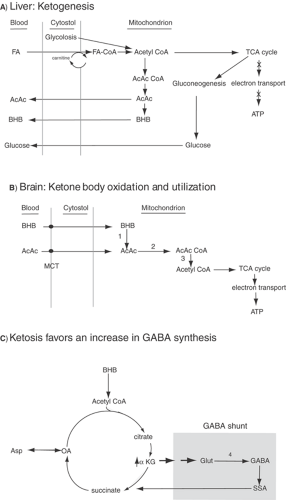 FIGURE 1. Ketogenesis and ketone body utilization by the liver and brain. A: Ketogenesis in the liver. Fatty acids (FAs) from the circulation enter hepatocytes, and then cross the inner mitochondrial membrane by either diffusion (short- and medium-chain FA) or via carnitine transport (long-chain FA). Under conditions of fasting or the ketogenic diet, carbohydrate substrate is lacking, so FAs are metabolized. Since oxaloacetate (a tricarboxylic acid [TCA] cycle intermediate) is diverted to gluconeogenesis, the TCA is not actively involved in energy generation (X’d arrows). Acetyl coenzyme A (CoA) is therefore not funneled into the TCA, but instead is used for ketone body synthesis (acetoacetate [AcAc], β-hydroxybutyrate [BHB]). Ketones are exported into the circulation, since the liver lacks the enzymes required to catabolize AcAc and BHB. B: In the brain, BHB and AcAc enter neurons via monocarboxylic acid transporters (MCTs). In mitochondria, BHB and AcAc are broken down (by enzymes 1, 2, and 3) into acetyl CoA molecules that can then enter the TCA cycle for energy production. C: TCA cycle and γ-aminobutyric acid (GABA) shunt. In ketosis, α-ketoglutarate is increased and GABA synthesis from glutamate (via enzyme 4) is favored.101 In addition, the GABA shunt, bypassing normal oxidative metabolism, facilitates GABA synthesis. ATP, adenosine triphosphate; AcCoA, acetyl CoA; citr, citrate; succ, succinic acid; OA, oxaloacetate; asp, aspartate; α-KG, α-ketoglutarate; glut, glutamate; SSA, succinic semialdehyde; 1, BHB dehydrogenase; 2, succinyl-CoA transferase; 3, acetoacetyl-CoA thiolase; 4, glutamic acid decarboxylase. |
The mechanism by which seizures induce neuronal damage includes mitochondrial dysfunction with the formation of injurious reactive oxygen species (ROS). The KD has recently been shown to increase the expression of brain mitochondrial uncoupling proteins,84 which act to reduce ROS formation. It was recently demonstrated that BHB and AcAc (in millimolar concentrations) prevented neuronal hyperexcitability induced by exogenous hydrogen peroxide administration in vitro, and that ketones were capable of significantly reducing ROS formation in isolated mitochondria.38 Additional support for the
involvement of the KD in enhancing brain metabolism comes from microarray studies of gene transcription. Of 42 gene transcripts upregulated by the KD, 39 encode mitochondrial proteins.6a Taken together, these observations suggest that the KD may protect against seizure activity (and may also be neuroprotective) through antioxidant mechanisms activated by fatty acids and ketones.
involvement of the KD in enhancing brain metabolism comes from microarray studies of gene transcription. Of 42 gene transcripts upregulated by the KD, 39 encode mitochondrial proteins.6a Taken together, these observations suggest that the KD may protect against seizure activity (and may also be neuroprotective) through antioxidant mechanisms activated by fatty acids and ketones.
Alterations in Neurotransmitters and Synaptic Transmission
The ketogenic diet may enhance the function of γ-aminobutyric acid (GABA), the main inhibitory neurotransmitter of the mammalian brain. GABA is synthesized from glutamate via the
action of glutamate decarboxylase (Fig. 1C). In pathways of ketone body metabolism (GABA shunt), glial conversion of glutamate to glutamine (a GABA precursor) is increased by ketosis.19,100 Enhanced GABA accumulation or function at the receptor level could lead to reduced cortical excitability. At the same time, the rate of glutamate conversion to aspartate, which has an excitatory role, is reduced.101 In ketosis, α-ketoglutarate, the precursor of glutamate, is increased, thereby enhancing the synthesis of GABA via the “GABA shunt” (Fig. 1C).19
action of glutamate decarboxylase (Fig. 1C). In pathways of ketone body metabolism (GABA shunt), glial conversion of glutamate to glutamine (a GABA precursor) is increased by ketosis.19,100 Enhanced GABA accumulation or function at the receptor level could lead to reduced cortical excitability. At the same time, the rate of glutamate conversion to aspartate, which has an excitatory role, is reduced.101 In ketosis, α-ketoglutarate, the precursor of glutamate, is increased, thereby enhancing the synthesis of GABA via the “GABA shunt” (Fig. 1C).19
Ketones mimic GABA structurally, so they could play a direct inhibitory role on cellular excitability by stimulating GABA receptors or enhancing their action. Ketogenic diet treatment (and calorie restriction) increases the expression of glutamic acid decarboxylase (GAD65 and GAD67) isoforms, providing a mechanism by which GABA synthesis might be increased on the KD.11 In a study utilizing a variety of seizure induction methods, the most consistent protection with the KD was found when seizures were induced by blocking GABA receptors (i.e., picrotoxin, bicuculline).4
Effects of Lipids and Polyunsaturated Fatty Acids
Fatty acids may be important in mediating an anticonvulsant effect. Typically, a KD includes mostly saturated fats of various chain lengths. No attempt is made to specify fat type or chain length in the KD formulation, except for the medium-chain-triglyceride diet, described above.34 In early clinical studies, there was a correlation between plasma lipids and seizure control in children with epilepsy treated with the diet.15 Plasma lipid levels peaked as KD-induced seizure control became maximal. In experimental studies, young rats fed a variety of different fat types developed seizure resistance independent of the level of ketosis but dependent upon qualitative differences (i.e., different sources of fats).16,53 In general, increasing the ketogenic ratio leads to improved KD efficacy.7,23
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


