Klippel-Feil Syndrome
Peter D. Pizzutillo
Martin J. Herman
HISTORICAL BACKGROUND
In 1912, Klippel and Feil published the clinical and anatomic description of L. Joseph, a 46-year-old tailor with a history of chronic abdominal pain and recurrent pleural effusions (1). His appearance was notable for an extremely short neck and a low hairline; physical examination revealed marked limitation of motion of his neck (Fig. 30.1). On December 13, 1911, L. Joseph was diagnosed with pleurisy, pulmonary congestion, and nephritis. He subsequently developed albuminuria and tachycardia and expired 1 month after presentation. Autopsy revealed an enlarged heart, abnormally formed kidneys, and abnormal spine. The occipital-cervical junction was anomalous and allowed motion in flexion and extension; no rotation was noted. The entire spinal column appeared to be composed of 12 vertebrae with the four most rostral cervical vertebrae included in a rudimentary fusion mass. Klippel and Feil interpreted their findings as congenital absence of the neck with ribs arising from the base of the skull and called this complex a “cervical thorax.” Dorsal kyphosis and dorsolumbar scoliosis were observed in the more caudal spine.
While prior scientific writings had reported patients with congenital abnormalities of the cervical spine, Klippel and Feil’s work constituted the first detailed description of congenital failure of segmentation of the cervical spine with associated conditions. Smith noted the earliest observation of congenital fusion of the cervical spine in an Egyptian mummy (circa 500 bc) (2,3). Autopsy of an anencephalic fetus by Haller revealed a cervical spine with only five vertebrae (2,4). Morgagni also described an anencephalic fetus with similar cervical anatomy and an adult with occipitoatlantal and C2-C3 fusions (2). Autopsy dissection of a 70-year-old male revealed congenital fusion from C2 to C7 (2). The earliest radiographic observation of congenital fusion of the cervical spine in a live patient was reported by Sick who also noted the coexistence of Sprengel’s deformity (2). The majority of early investigations and reports of Klippel-Feil syndrome (KFS) were based on autopsy evaluation of stillborn infants with multiple congenital anomalies, anencephaly, iniencephaly, occipitalization of the atlas, vertebral fusions, hemivertebrae, and platyspondyly (2,5, 6, 7, 8 and 9).
DEFINITION
KFS refers to a clinical entity characterized by individuals with a wide range of congenital anomalies who share the common feature of congenital failure of segmentation of cervical vertebrae (10) (Fig. 30.2). The classic triad described by Klippel and Feil of a short neck, low hairline, and limited cervical motion occurs in fewer than half of all individuals with KFS (2,11, 12 and 13) (Fig. 30.3). The cervical spine anomalies vary from simple two-segment failure of segmentation of vertebral elements to complex patterns of failure of segmentation of multiple vertebrae with anomalies of the occipitocervical junction that include occipitalization of the atlas and iniencephaly. The result is a spectrum of clinical expression (12,14).
ASSOCIATED CONDITIONS
Cosmetic concerns about the appearance of the neck and shoulders or limited neck motion prompt orthopaedic evaluation. Radiographs of the cervical spine display interesting anatomic variations from the normal spine and suggest the potential for the development of spinal instability and neurologic compromise. The incidence of instability or neurologic deficits is low in the child and adolescent, and evaluation of anomalies involving other organ systems is clinically more appropriate (14).
Urogenital anomalies occur in 30% of individuals with KFS and represent the most important nonskeletal association because of the potential for life-threatening renal disease. The most common renal anomaly is unilateral absence of a kidney (12,14—16). Other anomalies include absence of the hemitrigone, absence of both kidneys, absence of the ureter, ectopic kidney, horseshoe kidney, and hydronephrosis (17, 18 and 19). Genital anomalies are more common in the female and involve absence of the uterus, absence
of the vagina, absence of a Fallopian tube, ovarian agenesis, and duplication of the uterus (17,20, 21 and 22); undescended and dysplastic testicles have been reported in the male with KFS.
of the vagina, absence of a Fallopian tube, ovarian agenesis, and duplication of the uterus (17,20, 21 and 22); undescended and dysplastic testicles have been reported in the male with KFS.
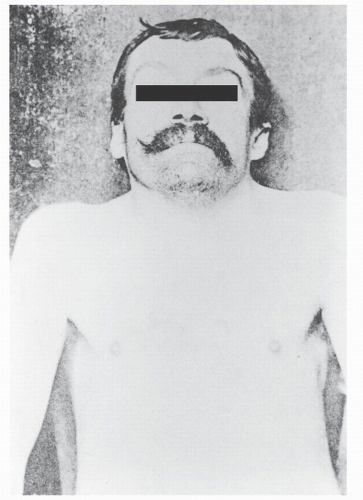 Figure 30.1. Photograph of L. Joseph with markedly shortened neck. (From Klippel and Feil, Un cas d’absence des vertebras cervicales avec cage thoracique remontant jusq’a du craine. Nouv Icon Salpetriere 1912;25:223.) |
Cardiovascular anomalies occur in 10% of individuals with KFS (2,23). Ventricular septal defects, alone or in combination with other cardiac anomalies, are most common. Mitral valve insufficiency, coarctation of the aorta, right-sided aorta patent ductus arteriosus, pulmonic stenosis, dextrocardia, truncus arteriosus, aplasia of the parietal pericardium, patent foramen ovale, single atrium, single ventricle, and bicuspid valve and atrial septal defects have been reported in KFS (2,23, 24, 25, 26, 27, 28 and 29). Thoracic anomalies include failure of lobe formation, ectopic lung, cervical ribs and absence or fusion of ribs, and deformities of the costovertebral joints (30,31).
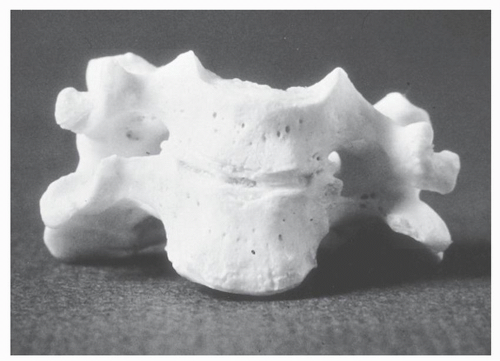 Figure 30.2. Anatomic specimen of single-level failure of segmentation. |
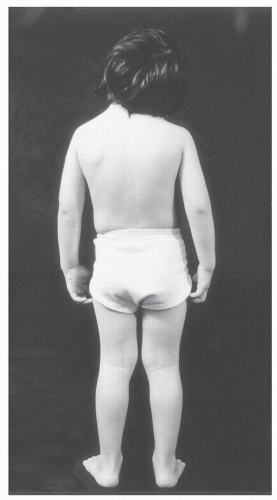 Figure 30.3. Patient with Klippel-Feil syndrome with short neck and low posterior hairline. |
Hearing deficits occur in 20% to 30% of individuals with KFS and may be sensorineural or conductive in nature with ankylosis of the ossicles, footplate fixation, or absent external auditory canal (2,14,25,32, 33, 34, 35 and 36). Early diagnosis of hearing loss is important to prevent delay in speech and language development. KFS has also been associated with defects in facial development with resultant cleft lip, cleft palate, high-arched palate, coloboma, ptosis, Duane
contracture lateral rectus palsy, and facial nerve palsy (24,37, 38, 39, 40 and 41).
contracture lateral rectus palsy, and facial nerve palsy (24,37, 38, 39, 40 and 41).
Synkinesia or mirror motion, cranial nerve anomalies, Chiari malformation, and syringomyelia have been reported in KFS. Iniencephaly is the result of failure of segmentation of cervical vertebrae, cervical dysraphism, and malformation of the base of the occiput with a massive foramen magnum that leaves the brainstem and cervical cord locked in extension with only the dura and skin as protective cover (42). These individuals exhibit severe neurologic deficits.
INCIDENCE
KFS is thought to occur in 1:40,000 to 42,000 births. Since few population screening studies exist, it is difficult to define its true incidence or prevalence. A female predominance of 3:2 has been reported. The heterogeneity of findings that have been included in “Klippel-Feil syndrome” is wide, and crossover with other syndromes such as Wildervanck, Rokitansky-Kuster-Hauser, and Goldenhar suggests that KFS may represent a spectrum of congenital cervical spinal anomalies with associated malformations (43,44).
ETIOLOGY/EMBRYOLOGY
During the 2nd and 3d week of fetal life, the development of the human blastema results in formation of the neural tube, which is surrounded by mesenchymal condensations. The condensations will become the future head, cardiac, paraxial, and lateral mesoderm. Paraxial mesoderm subdivides into somites proceeding from rostral to caudal. As the embryo matures, each somite further subdivides into three sections: the sclerotome, myotome, and dermatome. Resegmentation of the sclerotomes is followed by the union of the caudal half of one somite with the rostral half of the adjacent caudal somite resulting in the formation of a vertebral body (45).
Cross-sectional studies of the 28 to 30 day human embryo reveal marked proximity of the lower cervical somites to the paired dorsal aorta, the upper limb buds, the mesonephros, and the pronephric ducts (17,45). Pronephric ducts promote the formation of the mesonephros and ureteral buds that develop into the renal system. By 44 to 48 days of embryonic life, the pronephric ducts induce formation of Mullerian ducts, which give rise to ovaries, Fallopian tubes, uterus, and vagina.
KFS may result from disruption or mutations in the genes regulating the complicated sequence of somite segmentation and resegmentation of the occiput and cervical spine but has also been created in laboratory animals through teratogenic agents or artificial induction of maternal hypotension. Fetal alcohol syndrome has previously been loosely associated with segmentation defects of the cervical spine, but the association has had little supporting data (46,47). The associated anomalies of the urogenital, cardiac, and central nervous system suggest that alteration in gene regulation also influences the development of systems in proximity to the spine.
GENETICS OF KFS
In the latter half of the twentieth century, investigation of heredity in KFS involved observation of failure of segmentation of the cervical spine in family constellations. Reports of involved siblings with uninvolved parents and siblings of uninvolved consanguineous parents suggested autosomal recessive transmission (48). Da Silva’s report of 12 affected individuals in an inbred family of 59 individuals that suggested autosomal dominant transmission involved the clinical examination of 11 living members with radiographs of 8 of the 11. Genealogic data regarding other family members were derived by discussion and based on memory (49). Pizzutillo et al. (14) reported the radiographic evaluation of 49 patients with KFS in addition to 168 first-degree family members and noted only 4 of the 49 families with congenital failure of segmentation of the cervical spine. In each of the four families, the proband and the affected relative displayed simple single-level involvement. No patients with more complex patterns of cervical involvement had affected first-degree relatives.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


