Diagnostic Approach
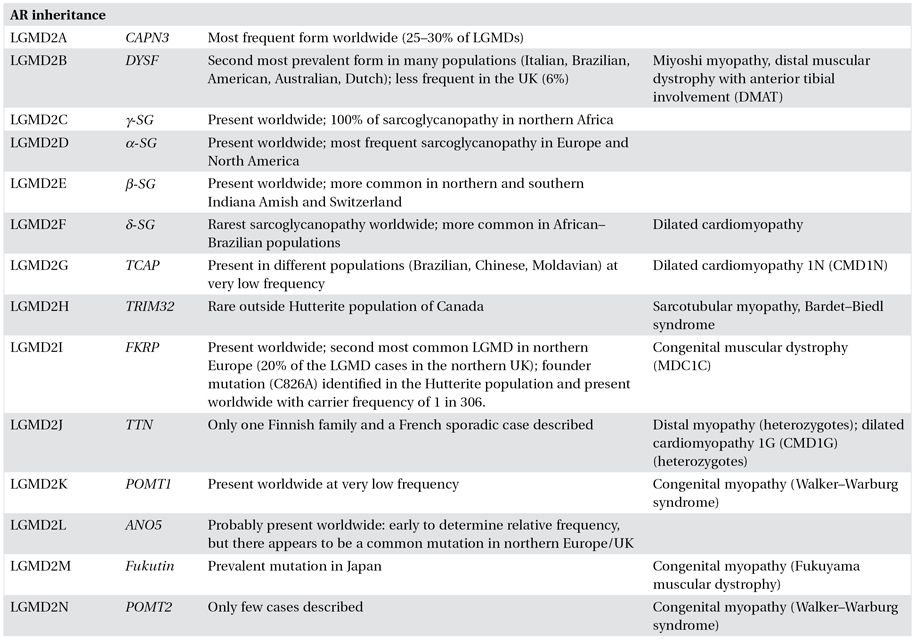
Although, per definition, LGMDs are characterized by a common clinical phenotype of predominantly proximal muscle weakness, there is enormous variability in clinical presentation, course and progression of symptoms, inheritance patterns, genes responsible, and proteins involved among the different LGMDs, and often also within the same form. Achieving a precise diagnosis of a particular type of LGMD can therefore be challenging and requires a comprehensive, multidisciplinary approach (Figures 8.1 and 8.2).
Figure 8.1. Diagnostic algorithm for achieving diagnosis in limb–girdle muscular dystrophies. CK, creatine Kinase; BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy; FSHD, facioscapulohumeral muscular dystrophy; LGMD limb–girdle muscular dystrophy; SMA, spinal muscular atrophy.
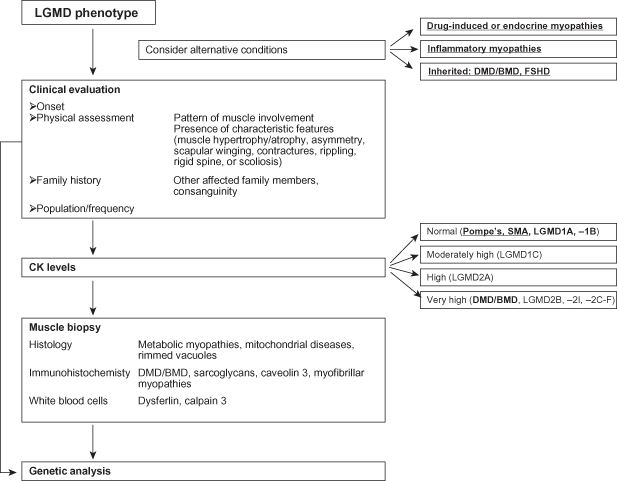
Figure 8.2. Diagnostic algorithm for autosomal dominant limb–girdle muscular dystrophies. AD, autosomal dominant; DM1, myotonic dystrophy type 1; FSHD, facioscapulohumeral muscular dystrophy; LGMD limb–girdle muscular dystrophy.
Clinical Approach
Age of onset, pattern of muscle involvement, additional clinical features (e.g. presence or absence of cardiac and/or respiratory involvement), and the presence of family history are all information that can be easily collected in clinic and are essential in directing the diagnosis and suggesting further investigations.
 tips and tricks
tips and tricks
- LGMDs often show a high inter- and intrafamiliar variability in clinical presentation and course.
- Clinical phenotypes may range from almost asymptomatic cases to severely affected patients within the same genetic condition and sometime the same gene defect.
- Some of the genes involved in LGMDs are also responsible for different phenotypes which might be completely distinct or represent a continuous clinical spectrum.
Onset and Medical History
The age of onset may vary between and within subtypes. Although a relatively well-defined age of onset has been reported in calpainopathies (onset in the early teens) and in dysferlinopathies (mean age of onset: 20 ± 5 years), a much wider age range has been described for most of the LGMDs. In general, dominant forms tend to present after the second decade of life and are usually slowly progressive, though even early childhood onset may be seen in laminopathy and LGMD1C. Early onset and relatively rapid progression are more common in sarcoglycanopathies, LGMD2I and the other LGMDs with abnormal α-dystroglycan glycosylation. A history of good sporting ability in childhood/early adulthood might suggest a diagnosis of LGMD2B or LGMD2L. Patients with dysferlinopathies are often misdiagnosed with an inflammatory myopathy due to a medical history of subacute onset of muscle pain and weakness not responding to steroids.
Muscle Assessment and Clinical Phenotype
A comprehensive muscle strength assessment helps in identifying specific patterns of muscle involvement, sometimes putting forward a specific diagnosis. Relative preservation of hip abductors with prominent scapular winging might suggest a diagnosis of LGMD2A, whereas prominent involvement of the posterior leg muscles and biceps brachii is characteristic of dysferlinopathies. Associated features, which need to be specifically sought, include muscle hypertrophy and atrophy, asymmetry of muscle weakness and wasting, scapular winging, rigid spine, scoliosis, contractures, and muscle rippling (Table 8.2).
Table 8.2. Clinical features of limb–girdle muscular dystrophies (LMGDs)
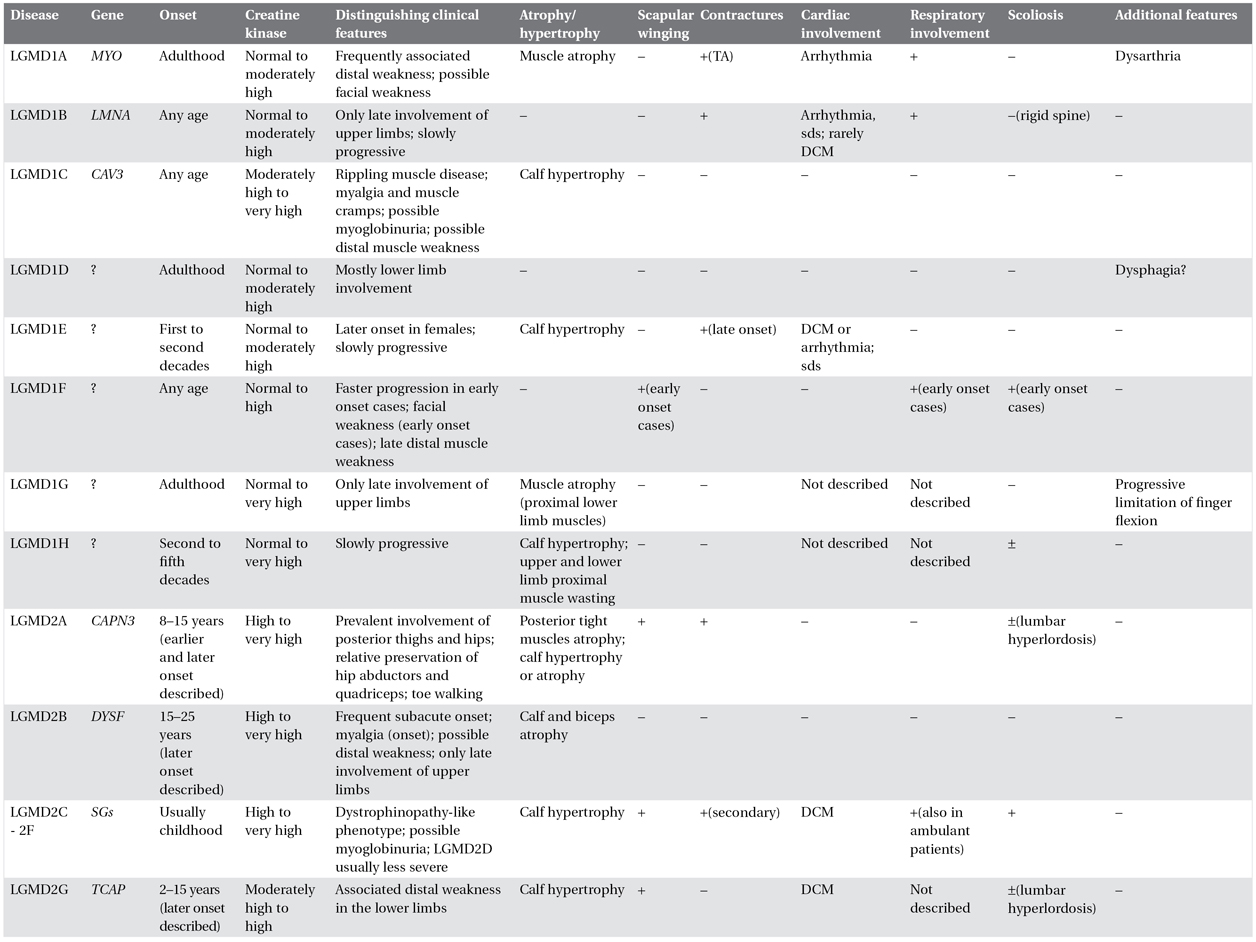
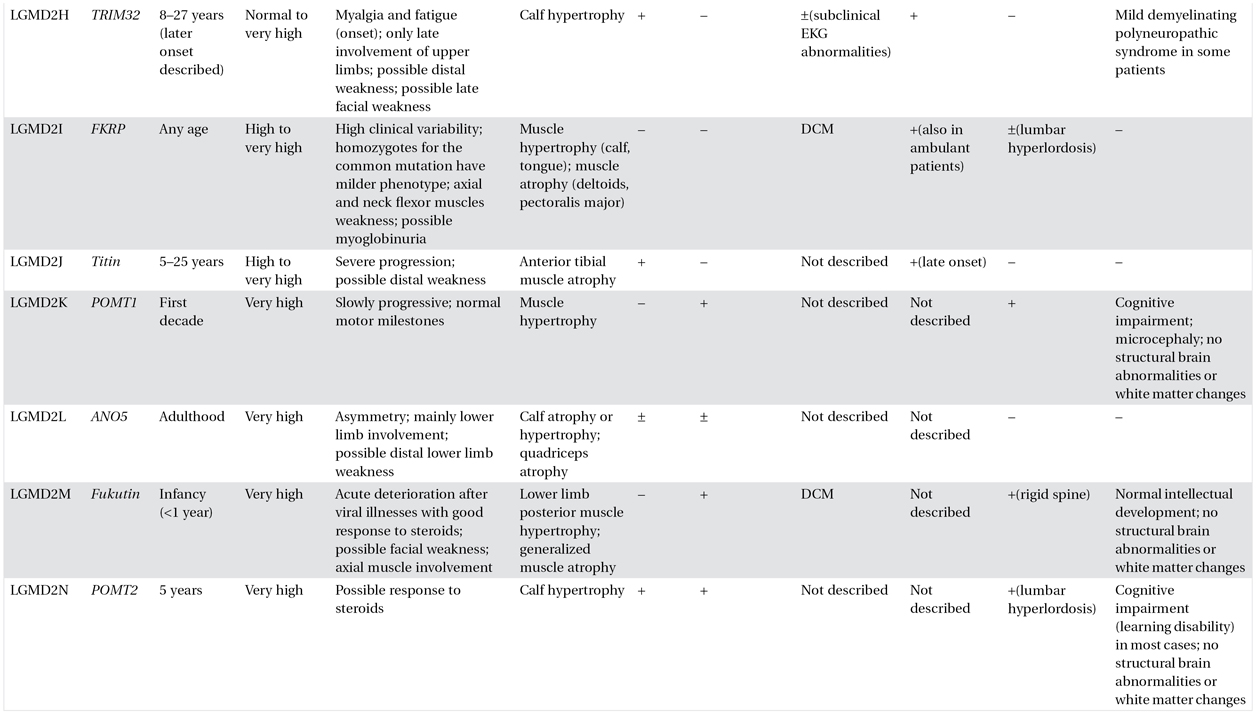
The table summarizes the most common clinical features of each form of LGMD. However, it is important to emphasize that exceptions to these general rules may be seen. This is due to a well-recognized inter- and intrafamilial variability among the different forms and to limited described cases of some forms. Therefore the reported clinical characteristics should be used as guidelines only, and caution should be exercised when approaching a patient with LGMD in both differential diagnosis and management.
TA: Achilles tendons; DCM: dilated cardiomyopathy; sds: sudden death syndrome.
CK: normal <200 IU/L; mildly elevated 200-500 IU/L; moderately high = 500–1000 IU/L; high = 1000–2000 IU/L; very high = >1000 IU/L.
Calf hypertrophy is a common clinical feature in LGMD1C, sarcoglycanopathies, and LGMD2I. However, dystrophinopathies (including carrier status) should also be suspected in the presence of prominent calf hypertrophy.
Focal muscle wasting and calf hypotrophy are most common in LGMD2A and -2B respectively. Lower limb distal muscle weakness might be observed in association with the LGMD phenotype in patients with LGMD1A, -2B, -2G, and -2L. Presence of muscle rippling and percussion-induced repetitive contractures are almost pathognomonic for LGMD1C, although there are also acquired causes of rippling muscle disease.
 tips and tricks
tips and tricks
- LGMD1B is the only LGMD that may result in neonatal hypotonia.
- Intellectual impairment is not characteristically seen in LGMDs and might suggest a differential diagnosis with dystrophinopathies.
- Normal sporting ability, subacute onset with muscle pain suggest a diagnosis of LGMD2B.
- Calf hypertrophy may frequently be observed in LGMD1C, -2C-F and -2I.
- Joint contractures are common in LGMD1B and may be observed in LGMD2A.
Family History
A detailed family history is essential when an inherited condition is suspected. Identifying a pattern of inheritance or the presence of consanguinity can significantly narrow the differential diagnosis and simplify the diagnostic workup. Questions about personal and family history should always extend beyond the neuromuscular system because, with several forms of LGMD, there may be multisystem disease. However, due to large intrafamilial variability and high occurrence of new mutations, especially in the autosomal dominant (AD) forms, a negative family history does not exclude the diagnosis of LGMD.
Epidemiology
It is now well recognized that LGMDs show variable patterns of disease frequency in different populations and therefore understanding the epidemiology of the condition in the population in question is helpful in outlining the differential diagnosis. The AD forms are rare, accounting for about 10% of cases. Calpainopathies seem to be the most common LGMD worldwide and LGMD2I appears to have a high prevalence in northern Europe. LGMD2L was only recently described and it seems to be relatively common in northern England and Germany. The availability of diagnostic genetic testing for anoctamin-5 may also confirm a high incidence of this form in other populations. The relative proportion of the other autosomal recessive (AR) forms varies from country to country (see Table 8.1).
Investigations
Serum Creatine Kinase
Serum creatine kinase (CK) represents a valuable, cheap, and noninvasive test, which helps in the differential diagnosis (see Table 8.2). As a rule of thumb, higher CK levels are observed in the AR compared with the AD forms, with the exception of LGMD1C. Within the AR LGMD, calpainopathies, dysferlinopathies, sarcoglycanopathies, and LGMD2I are usually associated with very to extremely high CK levels. Normal CK at onset of disease essentially excludes AR LGMD and is very unlikely even in the AD forms. It is important to note that, as with all forms of muscular dystrophy, CK levels fall with age and disease progression so in older patients CK levels may be normal or only minimally elevated.
Electrophysiology
Electromyography and nerve conduction studies are not likely to be of value in distinguishing the different forms of LGMD, but may be helpful in the differential diagnosis with other conditions such as SMA, myotonic dystrophies, and myasthenic syndromes.
Muscle Magnetic Resonance
Over the last few years, muscle magnetic resonance imaging (MRI) has been used to detect distinct patterns of muscle involvement in different neuromuscular conditions and may represent a promising noninvasive investigation in the diagnosis of the different forms of LGMD. However, reliable sequencing and protocols are so far available only in specialized centers and require highly qualified personnel with experience in muscle imaging and analysis. This expertise will hopefully become more widely available in the future.
Muscle Biopsy
Despite continuous progress toward understanding the molecular basis of LGMDs, a diagnostic approach exclusively based on genetic testing is often inefficient and too expensive to be applicable in clinical practice. Although the prospect of new generation sequencing and “LGMD chips” offers promise, for now such technologies are not widely applicable in the diagnostic setting. The muscle biopsy is therefore unavoidable in most cases and continues to be a highly informative and relatively economic diagnostic step for many forms of MD. No studies have compared open versus needle or conchotome biopsies; however, immunohistochemical and immunoblotting procedures often require a substantial sample to allow meaningful interpretation. Muscle MRI may be useful before biopsy to identify muscle with sufficient preservation to yield useful results, especially in advanced disease.
Basic histology is of little value in discriminating between the different LGMD forms but might be useful in the differential diagnosis. AD forms might show a nonspecific myopathic pattern without clear dystrophic abnormalities. Inflammatory infiltrates are common in dysferlin-opathies, whereas rimmed vacuoles might be seen in LGMD1A, -2G, and -2J although these changes are nonspecific. Muscle immunohistochemistry and immunoblotting are crucial in the diagnosis of the different forms of LGMD and in directing further genetic testing in most cases. If DMD/BMD has not been genetically excluded, demonstration of normal dystrophin expression is imperative, due to the higher incidence of dystrophinopathies compared with LGMDs. Immunoanalysis might show primary protein changes, clearly suggesting a specific genetic defect, such as caveolin 3 deficiency in LGMD1C. However, secondary protein deficiencies are commonly observed in some LGMDs, such as calpain 3 reduction in dysferlin deficiency and LGMD2J patients, and dystrophin reduction in all sarcoglycanopathies. Secondary reduction in caveolin 3 expression has also been described in patients with autoimmune rippling muscle disease. Moreover, normal protein expression on immunoanalysis can be observed in many LGMDs, including LGMD1A, -1B, and -2A (Table 8.3).
 caution!
caution!
Caution also needs to be taken in the presence of very specific clinical features as, even where protein testing is usually highly predictive, there may be cases with mutations and normal protein on antibody analysis.




