6 Low Grade Astrocytomas
Introduction
In the 1970s, 80% of nonglioblastoma gliomas were considered to be diffuse astrocytomas. The percentages of oligodendrogliomas and oligoastrocytomas have risen progressively subsequently; these tumors now account for 65% of grade II primary glioma diagnoses in the SEER database from 1994 to 2001.1 This increase reflects not a biological change, but, instead, an evolution in pathological interpretation prompted by the identification of a favorable-risk chemosensitive oligodendroglioma; this has created an “incentive” to find oligodendroglial elements and to determine whether there are deletions in 1p and 19q.
Diffuse Astrocytoma, WHO Grade II
PATHOLOGICAL AND MOLECULAR ASPECTS
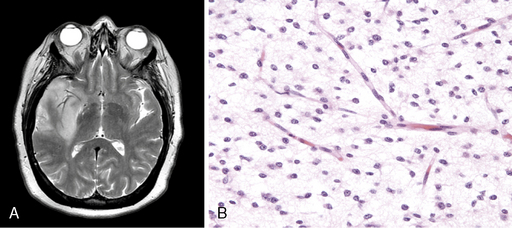
Diffuse astrocytoma is the most common astrocytoma. Because its cells produce neuroglial fibrils, this tumor is firm and rubbery. Its epicenter is typically in white matter. Cytologically, the more common fibrillary subtype is composed of atypical fibrillary astrocytes with hyperchromatic, elongated nuclei that appear “naked” within a fibrillary background or display discernable eosinophilic cytoplasmic processes. Fine and coarse neuroglial fibrils occupy the matrix and lie in random orientation among the cells from which they are derived (Figure 6-1). Mitotic activity is, by definition, absent in small biopsies and rare even in large resection specimens. Vasculature is inconspicuous, and necrosis is characteristically absent. Two pathological variants of the diffuse astrocytoma are the protoplasmic and the gemistocytic. The former is composed of cells resembling protoplasmic astrocytes. Protoplasmic tumors generally involve the cerebral cortex. Gemistocytic astrocytomas, by contrast, have cells with prominent eosinophilic cytoplasm that imparts a distended or globoid appearance and strong glial fibrillary acidic protein (GFAP) immunopositivity.
Unlike oligodendrogliomas, which frequently demonstrate losses of 1p and 19q and rarely have p53 mutations, many WHO grade II diffuse astrocytomas demonstrate loss of chromosome 17p, and roughly half harbor p53 mutations.2,3 The prognostic value of a p53 mutation is debated.2,4,5
Many WHO grade II diffuse astrocytomas harbor an alteration of a component of either the RB1 or the TP53 pathway, but few have alterations in both pathways. One study of 46 patients found a defect in the TP53 pathway in 70% and in the Rb pathway in 13%.6 The authors concluded that disruption of either the TP53 or the p14ARF pathway is frequent in low-grade astrocytomas and correlates with an unfavorable clinical course.
Even more common in WHO grade II diffuse astrocytomas is abnormal p53 protein immunoreactivity, which can occur even when mutations are not present.2 PDGFr-alpha overexpression is also common7 in these tumors.
A recent study comparing WHO grade II diffuse astrocytomas with primary and secondary GBMs using cDNA array analysis noted that WHO grade II diffuse astrocytomas had highly specific and homogenous expression profiles compared with the less specific and heterogenous profiles of primary GBMs; those of secondary GBM were intermediate.8 The distinguishing factor for primary GBMs was the upregulation of genes involved in angiogenesis. Such genes were not upregulated in WHO grade II diffuse astrocytomas, suggesting that angiogenesis is intimately linked with aggressive behavior.
CLINICAL ASPECTS
WHO grade II diffuse astrocytomas are generally slowly growing, locally infiltrative tumors in young or middle-aged adults.9,10 They rarely metastasize within11 or outside12 the CNS. They usually present as nonenhancing lesions on MRI (Figure 6-1). They are best viewed as “diffuse”; in other words, they are infiltrative, ill-defined tumors that can spread for many centimeters throughout the cortex and along white matter tracts, perivascular spaces, and periventricular ependyma in the form of secondary structures of Scherer, namely in perivascular and perineuronal locations. Regional heterogeneities in morphology, proliferative activity, ploidy and genetic aberrations are common.13 Although some patients survive for decades,14,15 these tumors have a propensity to evolve gradually into more aggressive anaplastic gliomas or GBMs,16,17 especially in older patients.16
Patients with supratentorial WHO grade II diffuse astrocytomas have a much higher incidence of seizures as a presenting symptom than do those with higher grade gliomas. Epilepsy is by far the most common and important symptom in WHO grade II diffuse astrocytomas. Seizures occur as a presenting symptom in approximately 50% of cases, and have a prevalence greater than 80%.18 Furthermore, patients with WHO grade II diffuse astrocytomas often suffer from neuropsychological and psychological problems that are aggravated by epilepsy and its treatment.19
The seizures originate not from the mass lesion but from adjacent brain tissue.20,21 Nevertheless, both radiation therapy22 and lesionectomy21,23,24 may significantly reduce or even eliminate medically refractory seizures.
Anticonvulsants, even multiple agents in combination, fail to control seizures in up to 50% of patients.19,25 They may also impair cognitive functioning.19 Since differences in efficacy are small, clinicians should choose an anticonvulsant with few side effects. Due to their effects on cytochrome P450 metabolism, the potential for interaction between anticonvulsants and other drugs (e.g., chemotherapeutics) can make it difficult to maintain adequate drug levels. Newer nonenzyme-inducing antiepileptic drugs such as levetiracetam and lamotrigine have equal efficacy, fewer side effects, and less frequent drug interactions than the more conventional agents, and are thus preferable to phenytoin and phenobarbital.
In a recent review of patients with intractable epilepsy and supratentorial tumors, including many with WHO grade II diffuse astrocytomas, improved seizure outcome was associated with a short duration of epilepsy before surgery, a single EEG focus, and the absence of hippocampal sclerosis or cortical dysplasia.26 The authors concluded that tumor resection was indicated for seizure control and prevention of malignant progression.
PROGNOSTIC FEATURES
Before CT scanning became readily available, reported median survival times for patients with WHO grade II diffuse astrocytomas were 5 years or less.27–30 More recent studies, however, show much longer median survival times, usually between 7 and 9 years, and 5- and 10-year survival rates range from 25% to 97% and 10% to 63% respectively.14,17,31–37 Two explanations have been offered for this longer reported survival: either patients are being diagnosed earlier or they are being more effectively treated. One recent study38 noted that use of newer imaging methods added no more than 6 months to the duration of survival following imaging diagnosis. Because radiation therapy has not been shown to improve survival, the authors speculated that longer survival resulted from more effective surgery, although they did note that macroscopically complete resections did not significantly improve survival duration as compared to less complete resections.
The outcome for patients with low-grade glioma (LGG) is intimately linked to anaplastic transformation. Pathologic progression of a diffuse astrocytoma to GBM produces an abrupt change in clinical behavior. The incidence of malignant transformation has been reported in various clinical studies to range from 13% to 86%.28,39–43 Because many of these series included previously treated patients, the natural history of this transformation is difficult to determine. A recent study reviewed 40 patients with progressive tumors who had undergone surgery but no further therapy for at least 3 months after the diagnosis of diffuse astrocytoma was made.44 Half had grade III or IV tumors when a second biopsy or operation was performed. Patients with progression had longer median times to reoperation (47 months vs. 22.5 months). The authors noted that complete resection delayed the time to reoperation but did not affect whether progression occurred.
The prognosis of adult patients with diffuse astrocytomas is largely determined by the inherent biology of the tumor27,32,37,43,45 and patient age.14,32,37,39,46,47 Adverse prognostic factors include poor performance status, tumor contrast enhancement, cognitive dysfunction, focal neurological deficits, and steroid dependency.14,27,32,37,39,43,46,48 In one study of several hundred patients accrued to two EORTC studies, multivariate analysis identified age greater than 40 years, astrocytoma histology subtype, largest diameter of tumor greater than 6 cm, tumor crossing the midline, and neurologic deficit present prior to surgery as the most important poor prognostic factors.49
TREATMENT ISSUES
Surgery provides tissue for histopathological analysis and can be helpful in relieving mass effect. However, because of the infiltrative nature of this tumor, gross total resection is often not possible. Nevertheless, most neurosurgeons favor attempting maximal resection at the time of surgery.49–52 However, it must be emphasized that these observations are difficult to interpret because of a bias in comparing these patients to those for whom it was not possible to do a maximal resection; therefore, the data should not be considered definitive and less extensive surgeries or even observation can still be justified.
Radiation therapy is also frequently advocated for diffuse astrocytomas, but the data supporting this approach are weak. An important study from the EORTC examined the effect of either immediate or delayed RT (54 Gy in 6 weeks) in a randomized study that included over 300 patients.53,54 Although immediate RT prolonged progression-free survival and seemed to result in better seizure control, no difference in survival was noted. The absence of a survival benefit suggested that although RT seemed to slow progression, it did not avert transformation into more aggressive GBMs. In addition, escalating the RT dose does not improve survival for these patients.46,50
Thus, there are no firm recommendations concerning RT in diffuse astrocytoma. Generally, clinicians use prognostic factors to choose patients who might benefit from immediate RT. These include persistence of significant disease-associated neurological dysfunction (including seizures), recurrent or progressive disease, age over 40, tumor size greater than 6 cm or crossing midline, and MIB proliferation index greater than 3%.49,50,55
There is no evidence that administration of chemotherapy prolongs survival in patients with diffuse astrocytoma.56 However, a recent report from the RTOG suggested some improvement in progression-free survival when RT was combined with procarbazine, CCNU, and vincristine (PCV), although 5-year survival was not significantly altered.57
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


