23 This chapter deals first with the lysosomal disorders that principally affect gray matter and then with those involving white matter (leukodystrophies). Lastly, the chapter covers the peroxisomal disorders, which include adrenoleukodystrophy. Other leukodystrophies are considered in Chapter 5. Chapter 5 also covers comparative aspects of all of the leukodystrophies, and an approach to their differential diagnosis. In the older patients neuronal storage of excessive lipofuscin is confined to the basal ganglia, brain stem, cerebellum, and spinal cord. In infantile GM2 gangliosidosis, ballooned neurons are found throughout the CNS and the peripheral nervous system. The foamy nerve cells stain strongly with Luxol fast blue and Sudan black, and in frozen sections the soluble ganglioside is periodic acid–Schiff (PAS)-positive (Fig. 23.1). Microglia are also PAS-positive, retaining stored material even in paraffin sections. Ultrastructural studies show membranous cytoplasmic bodies (MCBs) within neuronal somata. 23.1 GM2 gangliosidosis. Mild gyral atrophy is present in all three subtypes (Fig. 23.2a,b). Ballooned neurons with staining characteristics virtually identical to those seen in GM2 gangliosidosis are widespread in the cerebrum (Fig. 23.2c,d), brain stem, and spinal cord, and in autonomic ganglia (in subtypes 1 and 2). Ballooned neurons are confined to the striatum and pallidum in the adult form. Ultrastructural studies show membranous cytoplasmic bodies. 23.2 GM1 gangliosidosis. This group of disorders has a confusing set of eponyms (Table 23.1). Classification is further complicated by newer terminology related to the molecular genetics of the disorders. The classification used here combines clinical presentation, age of onset, pathology, and electrophysiology. The diagnosis is most readily obtainable for all forms except Kufs’ disease by cutting cryostat sections of a suction rectal biopsy to examine neurons and other cell types (i.e. smooth muscle, histiocytes, vascular endothelium) for the characteristic accumulations of autofluorescent ceroid lipofuscin, while ultrastructural examination for the various specific types of inclusions is most easily accomplished using buffy coat preparations of lymphocytes. In JNCL (CLN3) numerous vacuolated lymphocytes are demonstrated in the trails of a routine peripheral blood film, and in the right clinical setting this is virtually confirmatory. Skin biopsy is also routinely used: examination of eccrine but not apocrine glands is informative (if the biopsy is from the axilla note that many of the sweat glands will be of apocrine type). Cerebral atrophy is always present, but is at its most severe in the infantile form (Fig. 23.3a–c), manifesting as a walnut brain with shriveled cortex and rubbery white matter encased in a markedly thickened skull. Atrophy may also be considerable in infantile and juvenile Batten’s disease, and increases with the length of survival. In adult cases (Kufs’ disease), atrophy is more limited, and predominantly in frontal and cerebellar regions. 23.3 Batten’s disease. The stored material, which is insoluble and therefore readily detectable in paraffin as well as frozen sections, is widespread in the nervous system and in many other tissues. Its tinctorial properties vary slightly between the various subtypes of the disease (Table 23.2). In infantile Batten’s disease, storage is evident in CNS neurons, astrocytes, and macrophages, and in autonomic ganglia from an early stage, but neuronal loss is relatively subtle to begin with, becoming obvious after 2 years. By 4 years of age virtually all cortical neurons have disappeared, and there is dense astrocytic gliosis, and myelin loss. Some astrocytes contain storage material. In late-infantile and juvenile Batten’s disease (Fig. 23.3d,e) neuronal loss is less severe and myelin loss, if present, is slight. The rarity of adult cases and the accumulation of lipofuscin during normal aging have impeded the formulation of a consensus view of the histology of Kufs’ disease, although widespread storage is the principal element (Fig. 23.3f,g). Table 23.3 Classification of Niemann–Pick disease This classification incorporates the earlier four alphabetically defined groups. While hepatosplenomegaly is striking in both types, CNS abnormalities are not found in type B. In type A, cerebral atrophy may be slight or absent. Microscopically (Fig. 23.4), there is generalized enlargement of neurons and glia, and storage extends to white matter, which is demyelinated and gliotic. Gastrointestinal tract neuronal plexuses are also affected. Sudanophilic foamy histiocytes containing cholesterol esters, but not ballooned neurons, are numerous in the globus pallidus, substantia nigra, and dentate nucleus. Niemann–Pick cells (Fig. 23.4b) are present throughout the mononuclear phagocyte system, and can fill the alveolar spaces of the lungs. The lymphocytes of patients with type A Niemann–Pick disease contain cytoplasmic vacuoles, which are small and discrete. In contrast, in patients with type B there is minimal or no lymphocytic vacuolation. Bone marrow aspirates show collections of Niemann–Pick cells in patients with type A and in younger patients with type B. In older patients with type B disease there are fewer foamy Niemann–Pick cells, and more prominent ’sea-blue histiocytes’, in which the cytoplasm is filled with small granules that stain intensely blue with the Giemsa or Wright histochemical method. 23.4 Niemann–Pick disease type A.
Lysosomal and peroxisomal disorders
LYSOSOMAL DISORDERS
GM2 GANGLIOSIDOSIS
MACROSCOPIC APPEARANCES
MICROSCOPIC APPEARANCES
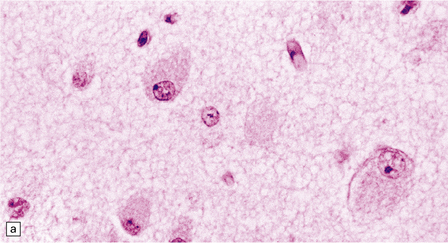
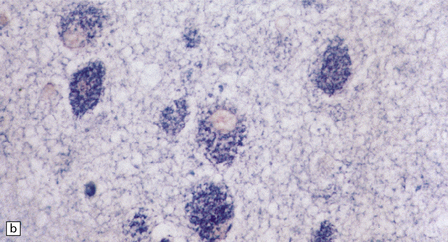
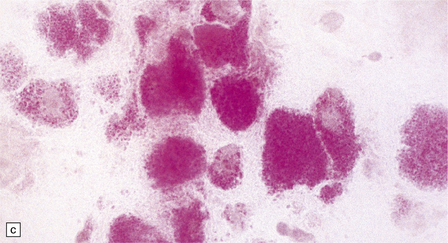
(a) Ballooned cortical neurons with foamy cytoplasm and marginated nuclei are negative with PAS in routine paraffin sections. (b) These neurons are, however, strongly stained with Luxol fast blue. (c) The stored ganglioside can be demonstrated in cryostat sections stained with PAS, in this example protected by celloidinization prior to staining.
GM1 GANGLIOSIDOSIS
MACROSCOPIC AND MICROSCOPIC APPEARANCES
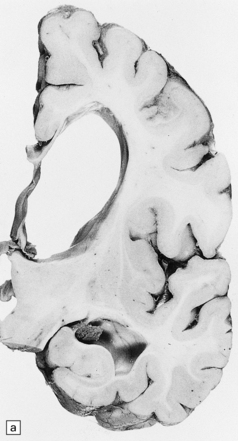
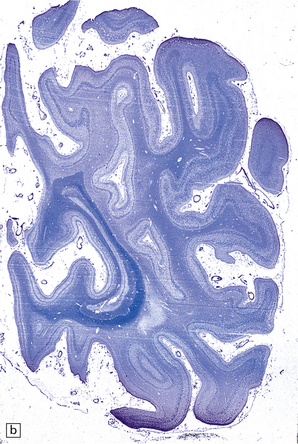
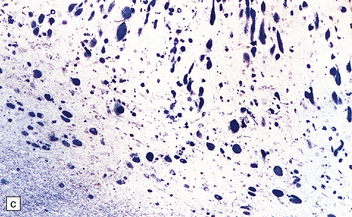
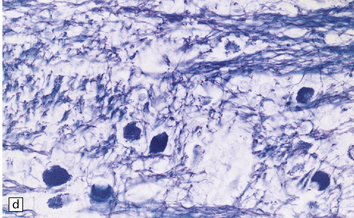
(a) Coronal section of cerebral hemisphere from a patient with the type 2 (late-infantile and juvenile) form of GM1 gangliosidosis, who died aged 10 years. There is mild cortical atrophy and ventricular dilatation. (b) Cortical atrophy and poverty of myelination are more evident in a stained section from the occipital lobe. (c) In the cerebral cortex there is extensive neuronal and glial storage. The granular storage bodies fill the neuronal cytoplasm and distend the proximal dendrites. Note also the myelin pallor. (d) Heavy neuronal storage is also present in the pontine nuclei.
BATTEN’S DISEASE, NEURONAL CEROID LIPOFUSCINOSIS (NCL, CLN)
MACROSCOPIC APPEARANCES
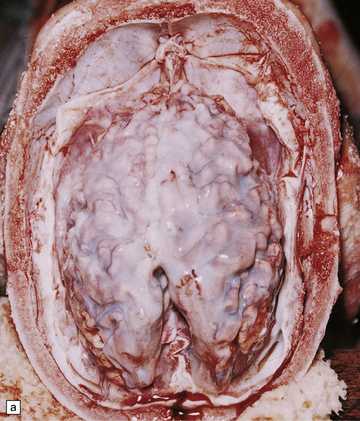
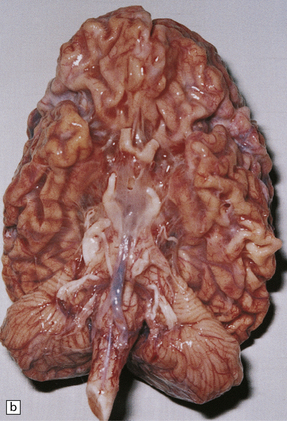
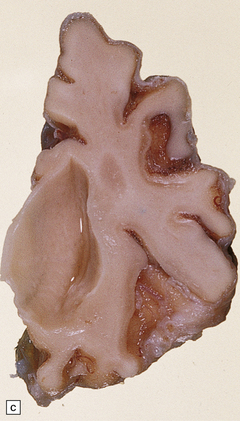
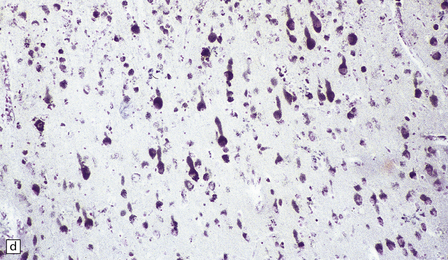
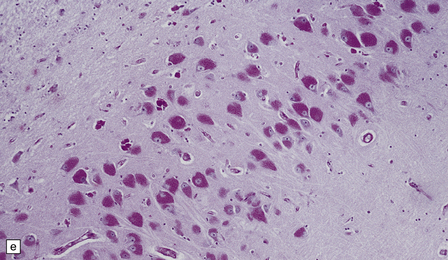
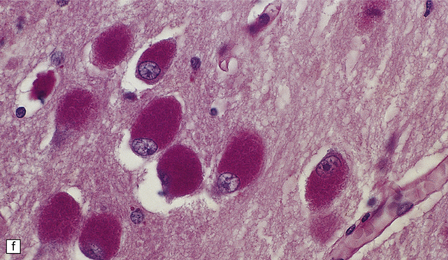
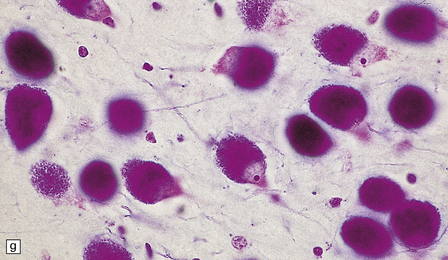
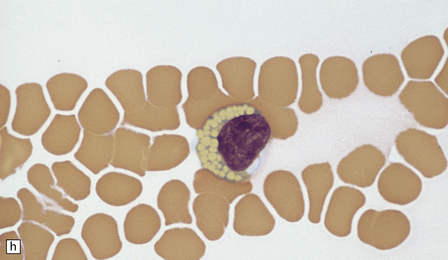
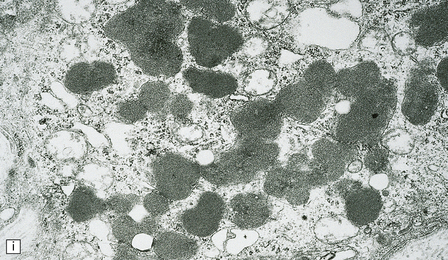
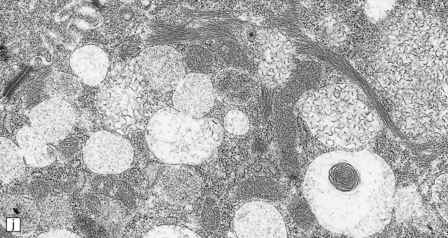
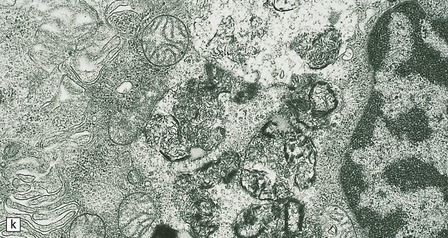
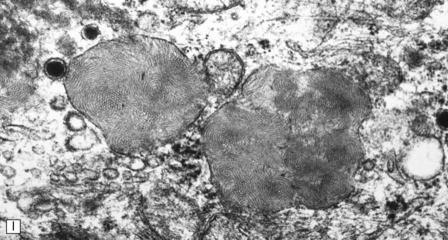
(a) Infantile Batten’s disease in a boy aged 9 years. The extremely atrophied brain (300 g weight) is covered by gelatinous leptomeninges and markedly thickened dura and surrounded by greatly thickened calvaria. (b) Viewed from below, the cerebral convolutional atrophy is marked and widespread, but the cerebellum and brain stem are relatively spared. (c) A coronal slice of frontal lobe shows a very thin cortical ribbon and tough rubbery white matter. (d) Juvenile Batten’s disease. Neuronal storage material reacts strongly with Sudan black in the cortex. (e) Juvenile Batten’s disease. Neuronal storage material reacts strongly with PAS in hippocampal pyramidal cells. (f) and (g) Kufs’ disease. In the rare adult form of NCL there is similar widespread neuronal storage material, which stains strongly with PAS. (h) Blood film of a patient with juvenile Batten’s disease showing a vacuolated lymphocyte with characteristic large uniform ‘bold’ vacuoles. A similar appearance is seen in GM1 gangliosidosis. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (i) Ultrastructural appearance in infantile Batten’s disease showing granular osmiophilic deposits in a neuron. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (j) Ultrastructural appearance in late-infantile Batten’s disease showing curvilinear bodies within a sweat gland epithelial cell. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (k) Ultrastructural appearance of a sweat gland epithelial cell containing mixed curvilinear and fingerprint bodies in juvenile Batten’s disease (similar in early juvenile and Finnish variant late-infantile Batten’s disease). (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (l) Fingerprint bodies in juvenile Batten’s disease.
MICROSCOPIC APPEARANCES
NIEMANN–PICK DISEASE
 Not sphingomyelinase deficient (Table 23.3).
Not sphingomyelinase deficient (Table 23.3).
Group I: sphingomyelinase deficient
Group II: not sphingomyelinase deficient
Type A: neurovisceral (infantile, juvenile, and adult)
Types C and D (Nova Scotia): neurovisceral
Type B: visceral only (infantile, juvenile, and adult)
Possible pure visceral form
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Niemann–Pick disease group I
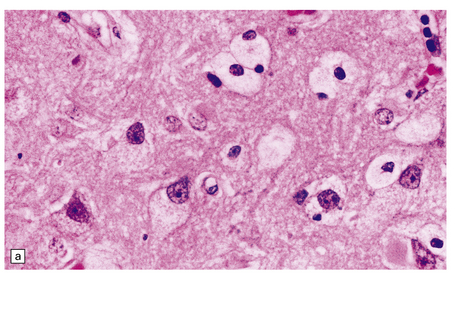
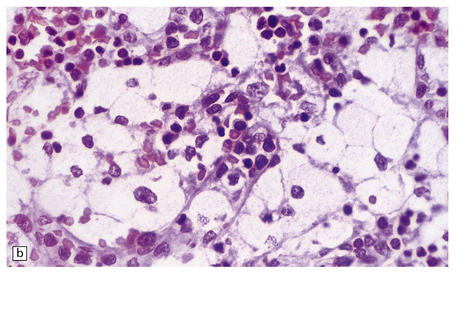
(a) Prominent neuronal and glial ballooning are seen in the cortex. (b) Large mulberry-like storage cells filling the red pulp of the spleen contain a single nucleus, uniform vacuoles, and occasional red cell debris.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Lysosomal and peroxisomal disorders
 GM2 GANGLIOSIDOSIS
GM2 GANGLIOSIDOSIS







 ETIOLOGY OF GM2 GANGLIOSIDOSIS
ETIOLOGY OF GM2 GANGLIOSIDOSIS









 GM1 GANGLIOSIDOSIS
GM1 GANGLIOSIDOSIS




 ETIOLOGY OF GM1 GANGLIOSIDOSIS
ETIOLOGY OF GM1 GANGLIOSIDOSIS

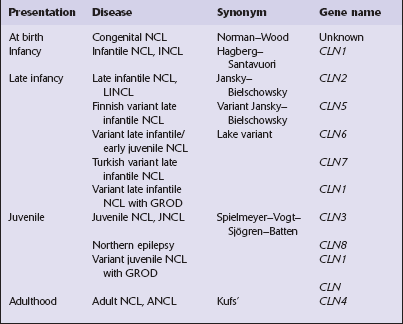
 ETIOLOGY OF BATTEN’S DISEASE
ETIOLOGY OF BATTEN’S DISEASE
 BATTEN’S DISEASE
BATTEN’S DISEASE













Only gold members can continue reading. Log In or Register to continue

