Medulloblastomas
Medulloblastomas are embryonal tumors arising from the cerebellum, initially described by Bailey and Cushing in 1925. Medulloblastoma is the most common malignant brain tumor of childhood, occurring at an incidence of 0.73 per 100,000 person-years and comprising approximately 25% of all pediatric brain tumors.1 Medulloblastoma can occur in all age groups; however, it is most common in children younger than 9 years of age, and the incidence decreases with increasing age thereafter. There exists a modest male preponderance in a ratio of 1.4:1, which is most notable in children younger than age 4. In the past, medulloblastoma was classified as a primitive neuroectodermal tumor (PNET); however, recent data have shown that medulloblastoma is biologically distinct from other intracranial PNETs, specifically supratentorial PNETs, pineoblastoma, and brainstem PNETs. As such, medulloblastoma is considered a disease distinct from other intracranial PNETs. Recent advances over the past 20 years have significantly improved our understanding of the biology of medulloblastoma, and advances in chemotherapy and radiation therapy have significantly improved outcomes.
40.1 Clinical Presentation
The clinical signs and symptoms of medulloblastoma are usually related to hydrocephalus secondary to obstruction of the fourth ventricle. Most tumors are diagnosed when they are quite large and obstruct the flow of cerebrospinal fluid (CSF) in the fourth ventricle; before this, most either are asymptomatic or cause subtle clinical symptoms. Almost all children with medulloblastoma present with symptoms of increased intracranial pressure, specifically early morning vomiting and headache. The common sequence of events is early morning headache relieved by vomiting and resolution of symptoms. As the tumor progresses, the diurnal variation of the headache and vomiting tends to become less pronounced, and these symptoms are more constant. Truncal ataxia and diplopia secondary to sixth nerve palsy are also common presenting features in addition to vomiting and headache.2 More lateral lesions may present with appendicular ataxia and occasionally focal weakness. Other neurologic symptoms localizing to the posterior fossa and brainstem can be present at diagnosis; however, they are much less common. Seizures rarely occur in children with medulloblastoma, and it is much more common for decerebrate posturing to be misinterpreted as tonic seizure. The time to diagnosis varies; however, children typically present acutely, and only rarely do children have a prediagnostic interval of longer than 2 months. It is not uncommon for children to undergo a gastrointestinal work-up before diagnosis. However, longer times to diagnosis do not correlate with reduced survival or worse neurologic outcome.3 The differential diagnosis for medulloblastoma at presentation includes ependymoma, pilocytic astrocytoma, atypical teratoid/rhabdoid tumor (AT/RT), and embryonal tumor with abundant neuropil and true rosettes (ETANTR). It is can be difficult to distinguish these entities based on clinical symptoms alone, although children with pilocytic astrocytomas tend to have a longer duration of symptoms, and children with ependymomas have a history of neck pain or stiffness with associated torticollis due to caudal invasion of the tumor through the foramen magnum.
Infants and very young children tend to have slightly different clinical presentations. Owing to their open sutures, infants present with macrocephaly and a head circumference that crosses percentiles. Infants with open sutures may present with irritability, lethargy, bulging fontanels, downward gaze due to pressure on the pretectum, and developmental arrest or regression. Very young children also tend to present with more catastrophic symptoms of hydrocephalus, such as apnea, bradycardia, and loss of consciousness, because of a more advanced stage of disease at diagnosis secondary to an increased tolerance of hydrocephalus. Adults commonly present with atypical symptoms and a longer prediagnostic interval because of a higher incidence of desmoplastic disease, which can arise in more lateral regions of the cerebellum. For example, patients who have cerebellopontine angle lesions present with vertigo, vomiting, nystagmus, and diplopia, which can be mistaken for vestibular canal symptomatology.
Metastatic disease is present in 40% of patients at diagnosis and is commonly asymptomatic; however, metastases may present with symptoms of nerve root involvement or spinal cord symptoms. Recurrent disease is rarely diagnosed clinically; rather, it is diagnosed on routine serial neuroimaging, and the likelihood of recurrence decreases with time.
Medulloblastoma can be associated with several familial cancer syndromes, specifically Gorlin syndrome, Turcot syndrome and Li-Fraumeni syndrome.4 Gorlin syndrome, also known as nevoid basal cell carcinoma syndrome, is present in 1 to 2% of patients with medulloblastoma and is an autosomal-dominant disorder associated with germline mutations in the PTCH gene on 9q31. The major criteria are multiple (more than two) basal cell carcinomas, jaw cysts (keratocysts) or bone cysts, palmar or plantar pits (more than three), early calcification of the falx cerebri, and a first-degree relative with Gorlin syndrome. Minor criteria include congenital skeletal abnormalities: specifically, rib abnormalities or fused vertebrae, macrocephaly, cardiac or ovarian fibroma, medulloblastoma, lymphomesenteric cysts, and various congenital malformations. Medulloblastoma in Gorlin syndrome usually develops before 2 years of age and is usually of desmoplastic histology and of the sonic hedgehog (SHH) subgroup. The outcome in these patients is more favorable than in those with sporadic medulloblastoma; however, basal cell carcinoma frequently develops in the irradiated skin, and these patients should be monitored on a long-term basis. Another germline mutation in the SHH pathway is hSUFU, which also leads to desmoplastic medulloblastoma but does not have the other manifestations seen in Gorlin syndrome.5,6 Turcot syndrome comprises distinct disorders: multiple colorectal neoplasms and tumors of the central nervous system (astrocytoma, medulloblastoma, pineoblastoma, ganglioglioma, ependymoma, glioblastoma). Medulloblastoma in patients with Turcot syndrome typically occurs before the age of 10 years and is not clinically different from sporadic disease. Turcot syndrome in medulloblastoma is associated with germline mutations in the adenomatous polyposis coli gene (APC) on 5q21. Li-Fraumeni syndrome is an autosomal-dominant disorder secondary to germline missense mutations in the TP53 gene on 17p13. Li-Fraumeni syndrome can lead to multiple different tumors, of which breast cancer, osteosarcoma, and brain tumors are the most common. Medulloblastoma occurs in approximately 5% of patients with Li-Fraumeni syndrome. Any patient with medulloblastoma and a strong family history of brain tumors or a previous history of an extraneural tumor warrants investigation for a germline TP53 mutation and should be monitored on a lifelong basis for the development of other tumors. More rare familial syndromes associated with medulloblastoma include Bloom syndrome and Fanconi anemia. Bloom syndrome is an autosomal-recessive disorder characterized by dwarfism, sun sensitivity, and a characteristic facial appearance. It is more common in Ashkenazi Jews and is secondary to a mutation in the BLM1 gene (RecQ protein-like-3) on 15q26.1. Fanconi anemia is an autosomal-recessive disorder that predisposes to bone marrow failure and leukemia and is secondary to several Fanconi anemia susceptibility genes.
40.2 Diagnostic Work-up
The initial diagnosis of medulloblastoma is usually made with noncontrast computed tomography (CT) followed by magnetic resonance (MR) imaging. On a noncontrast CT scan, a medulloblastoma is typically a hyperattenuating midline mass surrounded by vasogenic edema; it enhances homogeneously following the administration of contrast (▶ Fig. 40.1).7 The majority of lateral cerebellar medulloblastomas are of desmoplastic histology (▶ Fig. 40.2). Hydrocephalus is present on initial CT in over 95% of patients. The findings on CT are not specific for medulloblastoma and cannot reliably be distinguished from those of ependymomaor AT/RT. MR imaging with gadolinium enhancement is the preferred method for the initial evaluation of posterior fossa tumors. Younger children require general anesthesia for image acquisition, and care should be taken in these instances because there is a risk for respiratory depression. When sedation is indicated, it should be administered by a pediatric anesthesiologist. On T1-weighted MR imaging, medulloblastomas are hypointense or isointense relative to gray matter and become hyperintense with the administration of gadolinium. On T2-weighted imaging, the signal is variable and can be hyperintense to hypointense relative to gray matter. Diffusion-weighted imaging can be very useful in discriminating medulloblastoma from ependymoma or astrocytoma (▶ Fig. 40.3). Because medulloblastomas consist of dense small round cells with minimal cytoplasm and reduced free water, they exhibit diffusion restriction with a reduced apparent diffusion coefficient. This is in contrast to ependymomas and pilocytic astrocytomas, which typically do not have restricted diffusion.8 AT/RTs also display restricted diffusion; however, young age of the patient, cerebellopontine angle involvement, and intratumoral hemorrhage can help distinguish AT/RTs from medulloblastomas radiologically.9 When feasible, preoperative imaging of the entire craniospinal axis is desirable for adequate staging of disease because 40% of medulloblastomas are metastatic at diagnosis (▶ Fig. 40.4). However, imaging of the entire craniospinal axis should be performed 2 weeks or later postoperatively for full staging to avoid the possibility of false-positives due to debris and blood products.10 Cranial MR imaging should also be repeated within 72 hours postoperatively or after 2 weeks postoperatively because residual tumor of more than 1.5 cm2 may be correlated with a higher risk for disease.11 Cranial MR imaging performed between 3 and 14 days postoperatively can be falsely positive because of the presence of blood products and debris. Ultimately, pathologic diagnosis is required for a firm diagnosis.
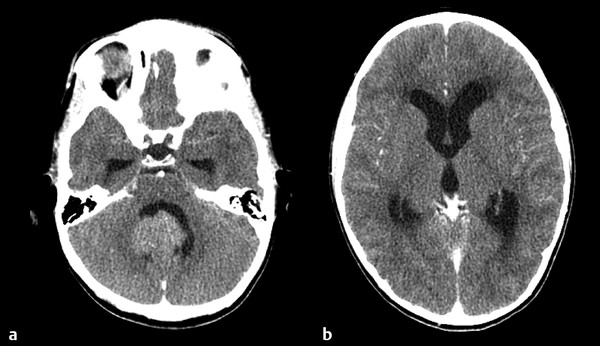
Fig. 40.1 Contrast-enhanced computed tomographic scan through the (a) posterior fossa and (b) third ventricle. Note the significant triventricular hydrocephalus at diagnosis.
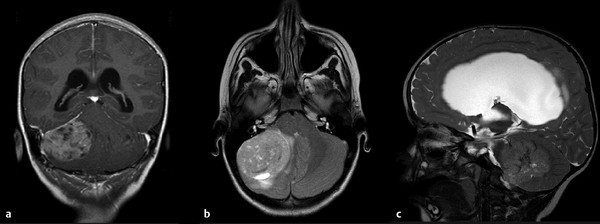
Fig. 40.2 (a) Coronal T1-weighted gadolinium-enhanced MRI. (b) Axial T2-weighted MRI. (c) Sagittal T2-weighted MRI showing a lateral desmoplatic tumor, commonly seen with the sonic hedgehog subgroup.
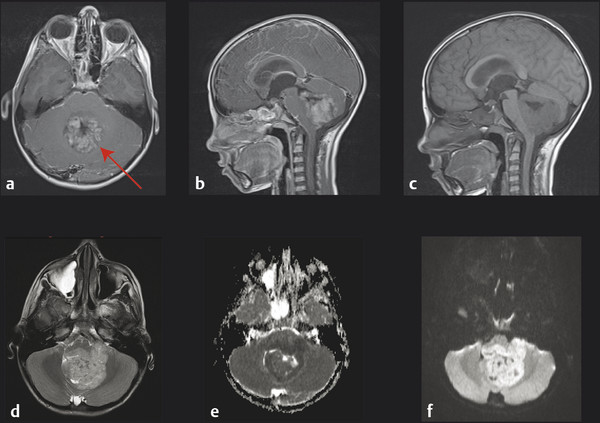
Fig. 40.3 Magnetic resonance (MR) image at diagnosis of a midline medulloblastoma. (a) Axial and (b) sagittal T1-weighted gadolinium-enhanced MR images. Note the homogeneous enhancement following contrast administration. (c) Sagittal T1-weighted unenhanced MR image. (d) Axial T2-weighted MR image (note the peritumoral edema). (e) Axial apparent diffusion coefficient (ADC) map and (f) axial diffusion-weighted Image (b1000). Note the restricted diffusion observed throughout the tumor.
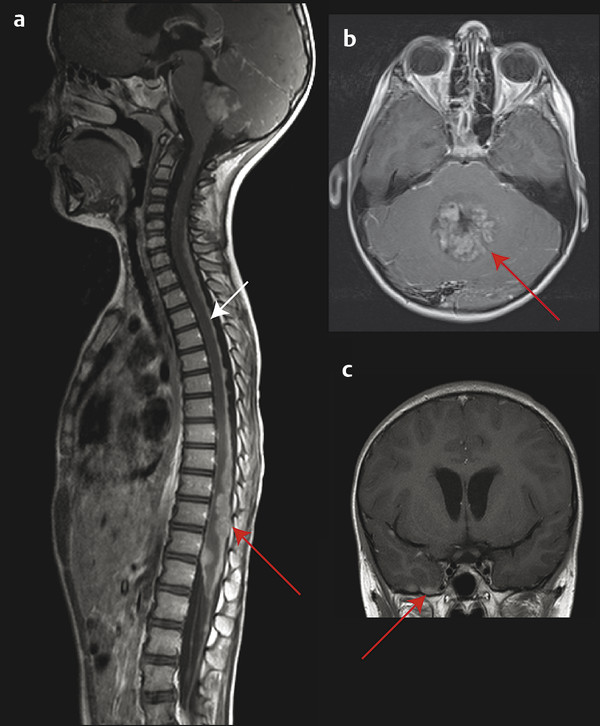
Fig. 40.4 Metastatic medulloblastoma. (a) Sagittal T1-weighted gadolinium-enhanced magnetic resonance (MR) image of the entire spinal cord showing diffuse nodular (red arrow) leptomeningeal metastases throughout the entire spine. (b) Sagittal T1-weighted gadolinium-enhanced MR image of the lower thoracic and lumbar spinal cord showing a classic “sugar coating” pattern of laminar metastases (white arrow) covering the spinal leptomeninges. (c) Coronal T1-weighted gadolinium-enhanced MR image showing supratentorial nodular metastases (red arrow) along the inferior surface of the right temporal lobe.
Lumbar CSF sampling is also warranted in every patient with a new diagnosis of medulloblastoma. Lumbar sampling is preferred over ventricular sampling at surgery or through a shunt because lumbar CSF sampling is more sensitive than ventricular sampling.12,13 Ideally, both ventricular CSF sampling at surgery and lumbar CSF sampling 2 weeks postoperatively should be obtained because there are cases of discordance between the two sites, and positive ventricular CSF cytology intraoperatively has been associated with a poorer outcome.13,14 Spinal MR imaging is not a substitute for lumbar CSF sampling and vice versa because up to 18% of cases of leptomeningeal disease can be missed if only one of the two modalities is used.15
There are a paucity of data regarding the role of bone marrow sampling at diagnosis; however, routine extraneural staging is not warranted because extra neural disease is extremely uncommon at diagnosis. If there is suspicion of extraneural disease, FDG-PET (fluorodeoxyglucose F 18 positron emission tomography) and bone marrow biopsy are the preferred modalities. Bone marrow sampling should be considered if the child will be undergoing autologous stem cell harvest as part of the treatment regimen. Extraneural disease is more commonly present at recurrence, particularly very late recurrences, and can be present in the absence of intracranial disease.16 Hearing and creatinine clearance should be evaluated postoperatively in all patients before the initiation of either radiation therapy or chemotherapy because both platinum-based chemotherapy and radiotherapy are ototoxic and nephrotoxic.
40.3 Hydrocephalus
The majority of patients with medulloblastoma present with hydrocephalus due to obstruction of the fourth ventricle. As such, the management of hydrocephalus is usually the first intervention. Most patients can be managed with preoperative dexamethasone (0.45 mg/kg per day divided into three doses, with an initial dose of 0.45 mg/kg), which results in significant alleviation of the symptoms and a reduction in vomiting. Placement of an extraventricular drain (EVD) preoperatively is occasionally necessary; however, in instances in which hydrocephalus is resulting in cardiopulmonary instability and/or in cases of impending herniation, it is warranted as a tempering measure until resection can take place. When an EVD is placed, consideration must be given to the possibility of upward herniation, and the rate and quantity of CSF drainage must be carefully monitored. The height of an EVD can be gradually increased in the postoperative period, and in most cases the EVD can be successfully removed within a week to 10 days postoperatively. From 10 to 40% of children require permanent CSF diversion postoperatively, and as such, routine ventriculoperitoneal shunting is not indicated perioperatively. The need for shunting is greatest in young children and in those with very large ventricles at diagnosis, more extensive tumors, and metastatic disease.17 CSF diversion is rarely required in children older than age 10. When persistent hydrocephalus is present, either ventriculoperitoneal shunting or endoscopic third ventriculostomy can be considered.18 Several series have reported high success rates with endoscopic third ventriculostomy for persistent hydrocephalus, and it should be considered in patients requiring postoperative shunting who are suitable candidates for this procedure. There is limited evidence for the use of routine endoscopic third ventriculostomy in all patients because a significant proportion of patients do not have persistent hydrocephalus.18 Rare cases of peritoneal seeding of medulloblastoma through ventriculoperitoneal shunts have been described.19
40.4 Tumor Resection
Gross total resection of the primary posterior fossa mass through a suboccipital craniotomy is the most common initial treatment for medulloblastoma. The goal of surgical resection is to achieve a gross total resection or, if this is not possible, to resect as much of the tumor as is safely possible. There is seldom any role for a limited biopsy. The craniotomy is performed with the patient in the prone position and the neck flexed. Once the superficial tissues and muscles are dissected, a portion of the occipital bone and the lamina of C1 are removed. The dura covering the cerebellum is then opened, and the two hemispheres are retracted through either the split vermis or a telovelar approach. Although medulloblastoma rarely invades the floor of the fourth ventricle, it is important to visualize the floor of the fourth ventricle at the onset of the surgery to ensure that the resection is not carried into the brainstem. Severe cranial nerve palsies can result if the surgeon aggressively resects tumor that invades the brainstem, and this is never indicated because the tumor is sensitive to both chemotherapy and radiation, and residual brainstem disease likely does not significantly alter outcome.11 If the tumor invades the cerebellopontine angle, then the use of intraoperative brainstem auditory evoked potentials can be considered. Medulloblastomas can be fairly vascular, leading to considerable hemorrhage intraoperatively, and care should be taken to prevent blood loss, particularly in young children. Dural closure should be achieved in a watertight fashion and the bone replaced after the procedure has been performed. Postoperative MR imaging should be performed to determine the extent of resection within 72 hours of surgery so as to avoid obscuration by blood products and gliosis. Residual disease over 1.5 cm2 is possibly associated with poorer outcome, and repeated resection for large residual tumor should be considered unless the surgeon stopped the initial resection because of excessive vascularity or invasion of critical structures.11
40.5 Pathology
The definitive diagnosis of medulloblastoma is made on microscopic pathologic examination. On gross examination, medulloblastoma appears as a pinkish gray to purple mass with a clear interface separating it from normal cerebellum. In the operating room, it also appears as a highly vascular lesion owing to its blood supply principally from the posterior inferior cerebellar artery. Leptomeningeal metastases may be seen at the time of surgical resection as well, which appear as a whitish coating that resembles icing sugar. On microscopic examination, medulloblastoma appears as a “small round blue cell tumor” and is morphologically indistinguishable from supratentorial PNETs or pineoblastomas. Medulloblastomas are highly cellular and the cells are densely packed, with prominent nuclei and scant cytoplasm. A type of pseudorosette may be seen (Homer-Wright rosette) in which differentiated cells surround the neuropil. Histologically, there are three main types of medulloblastoma: (1) classic, (2) desmoplastic, and (3) large cell/anaplastic. Classic histology is by far the most common histology (> 70%) and consists of sheets of small blue cells with densely packed isomorphic nuclei and a high proliferative index. Desmoplastic histology is more common in infants and adults and is most commonly associated with SHH pathway activation. Desmoplastic histology is characterized by nodules with a paucity of tumor cells surrounded by cells with a higher density, more pleotrophic nuclei, and densely packed reticulin fibers. Within the nodules, there is increased synaptophysin staining, and outside the nodules, there is a higher Ki-67 proliferation index, and the tumor cells are GFAP (glial fibrillary acidic protein)–positive. There is no consensus regarding how much desmoplasia is required to classify a tumor as desmoplastic. A variant of desmoplastic histology termed medulloblastoma with extensive nodularity (MBEN) is characterized by extensive nodularity and neuronal differentiation. The desmoplastic and MBEN variants in infants signified a favorable prognosis in several studies.20 The large cell/anaplastic variant is aggressive and characterized histologically by large, irregular, and pleomorphic nuclei; circumscribed foci of necrosis; and apoptotic bodies. The large cell variant tends to have prominent nucleoli. The large cell/anaplastic variant accounts for approximately 4%, is more common in the group 3 molecular subgroup, is commonly associated with bulky spinal metastases at diagnosis, and has a poor prognosis. Pathologic variants include the medullomyoblastoma, which is characterized by a striated muscle component, and the melanotic medulloblastoma, which is characterized by a pigmented epithelial component forming tubules or clusters. Medullomyoblastomas are more commonly associated with isochromosome 17q, have frequent MYC amplification, and tend to be of large cell/anaplastic histology; however, because of a paucity of multicenter studies, their prognosis is indeterminate.21 Melanotic medulloblastoma is very rare, and its prognostic significance is unclear.
The pathologic differential diagnosis for medulloblastoma includes AT/RTs and ETANTRs. AT/RTs are most common in young infants and should be considered in any child younger than age 5 with medulloblastoma, or when a rhabdoid component is present on histologic examination. Molecularly, AT/RTs are characterized by loss of INI1/hSNF5. Therefore, INI1 immunoreactivity should be assessed in all infants in whom medulloblastoma is diagnosed; in AT/RTs, staining for INI1 will be negative in the tumor but positive in blood vessels, and in medulloblastoma, staining will be positive in both tumor and blood vessels. ETANTRs are rare tumors that can occur in any location, including the posterior fossa, and were previously called ependymoblastomas. ETANTRs are characterized molecularly by focal amplification of a microRNA cluster at the 19q13.42 locus and should be considered when the pathologic features include ependymoblastic rosettes and neuronal differentiation on a neuropil background.22 ETANTRs are found in young infants and portend a poor prognosis.
The advent of integrated genomics over the past 20 years has significantly advanced our understanding of the biology of medulloblastoma. Several integrated genomic studies during the past 6 years have revealed that medulloblastoma comprises at least four variants, which are transcriptionally, genetically, and clinically distinct.23–25 These four subgroups are termed WNT, sonic hedgehog (SHH), group 3, and group 4 (▶ Fig. 40.5). The four molecular subgroups have significant prognostic value and predict survival independently of the presence of metastases or unfavorable histology. The WNT subgroup is characterized by activation of the WNT pathway, and tumors commonly harbor mutations in the β-catenin gene (CTNNB1). Patients with WNTactivated tumors, which occur primarily outside the infant age group, tend to have a very favorable prognosis.24,26 Patients with WNTactivated tumors are being considered for de-escalation of therapy in future clinical trial designs. The SHH subgroup is characterized by activation of the SHH pathway and is more common in infants with desmoplastic tumors and in adults; however, it can occur in all age groups and is associated with all histological variants. Infants with desmoplastic SHH-activated tumors have a favorable prognosis; however, other SHH-activated tumors have an intermediate prognosis. SHH pathway inhibitors such as smoothened inhibitors are currently in clinical trials and hold promise for personalized therapy in this subgroup of patients.27 Group 3 medulloblastomas have a poor prognosis and are commonly associated with metastatic disease. MYC amplification is common in group 3 medulloblastomas, and the survival rate in these patients is dismal. Group 4 medulloblastomas have an intermediate prognosis and are commonly associated with isochromosome 17q and MYCN amplification. A molecular subgroup can be determined through several methods, including immunohistochemistry with a four-antibody method (DKK1 for WNT, SFRP1 for SHH, NPR3 for group 3, and KCNA1 for group 4) and gene expression profiling through either a whole transcriptome or a limited-gene approach.23,28 WNT subgroup medulloblastomas can also be diagnosed through nuclear immunoreactivity of β-catenin.29 Other immunohistochemical approaches for the diagnosis of the four subgroups include GAB1 immunoreactivity for SHH and negativity for YAP1 and filamin A for non-SHH/WNT. Markers of a poor prognosis for non-SHH/WNT subgroups include FSTL5 overexpression, MYC amplification, MYCN amplification, and gain of chromosome 17q.24,30 TP53 mutations have been suggested to portend a poor prognosis; however, the literature surrounding this is conflicting, including the observation that patients with TP53 mutations in WNT-activated tumors have a good survival.31,32
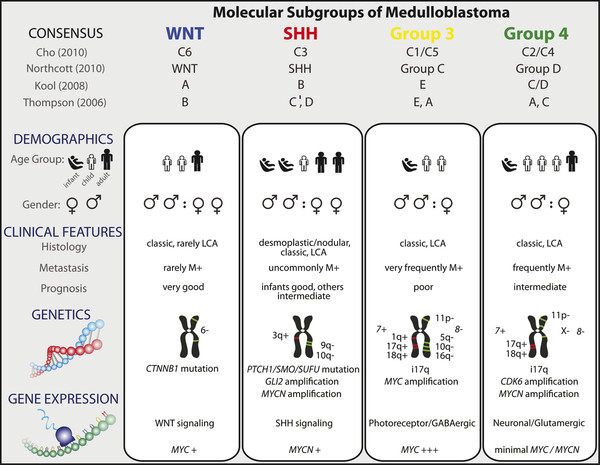
Fig. 40.5 Consensus molecular subgroups of medulloblastoma. Comparison of the various subgroups of medulloblastoma, including patient demographics, common genetic changes, and outcome. GABA, γ-aminobutyric acid; LCA, large cell/anaplastic; SHH, sonic hedgehog.
(Used with permission from Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123:467.)









