TABLE 7.1 Most Common Presentations of Neurologic Metabolic Diseases by Age | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
TABLE 7.2 Basic Initial Workup for Suspected Metabolic Disorder | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
TABLE 7.3 Advanced Workup for Suspected Metabolic Disorder | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
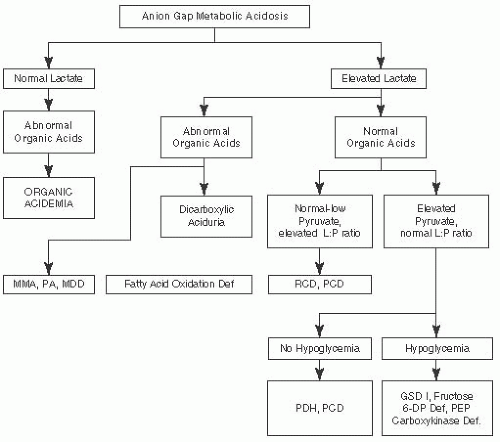 FIGURE 7.1 Metabolic Emergencies: Anion Gap Metabolic Acidosis. Adapted from Saudubray JM, Charpentier C. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York, NY: McGraw-Hill; 2000 and Fenichel GM. Clinical Pediatric Neurology. 6th ed. Philadelphia, PA: Saunders, Elsevier; 2009. |
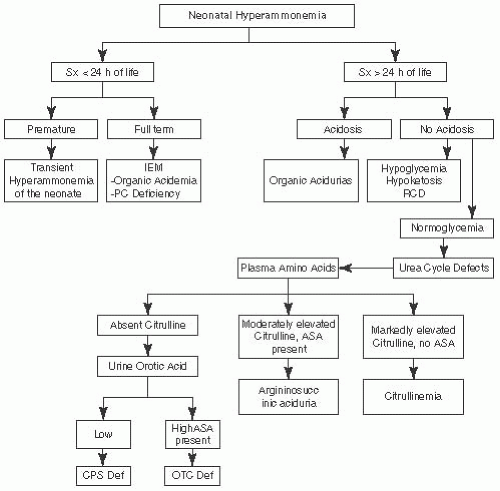 FIGURE 7.2 Metabolic Emergencies: Neonatal Hyperammonemia. Adapted from Saudubray JM, Charpentier C. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York, NY: McGraw-Hill; 2000 and Fenichel GM. Clinical Pediatric Neurology. 6th ed. Philadephia, PA: Saunders, Elsevier; 2009. |
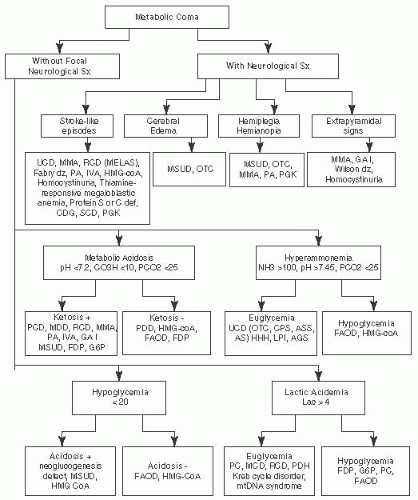 FIGURE 7.3 Metabolic Emergencies: Metabolic Coma. Adapted from Saudubray JM, Charpentier C. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York, NY: McGraw-Hill; 2000 and Fenichel GM. Clinical Pediatric Neurology. 6th ed. Philadelphia, PA: Saunders, Elsevier; 2009. |
TABLE 7.4 Differential Diagnosis of Lactic Acidosis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Hyperammonemia: immediate hemodialysis (HD) for coma, ventilator dependency, or signs of cerebral edema
Acidosis: give bicarbonate w/ frequent ABGs, severely acidotic → HD
Organic acidemia: Vitamin B12 (1 mg) IM for B12 responsive form of methylmalonic acidemia. Biotin (10 mg) PO or NGT for biotin-responsive carboxylase deficiency. May need HD or peritoneal dialysis if coma
Urea cycle defect: 6 cc/kg of 10% arginine HCL IV over 90 min
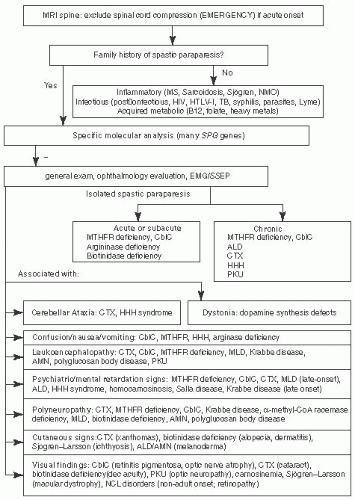 FIGURE 7.4 Progressive Spastic Paraparesis Associated with Metabolic Disorders. Adapted from Sedel F, Fontaine B, Saudubray JM, et al. Hereditary spastic paraparesis in adults associated with inborn errors of metabolism: a diagnostic approach. J Inherit Metab Dis. 2007;30(6):855-864, with permission. |
IV glucose (calories needed to keep euglycemia) D10 150 cc/kg/d w/ electrolytes
STOP protein (after 24-48 h restart essential amino acids), IV lipids should be given for urea cycle defects
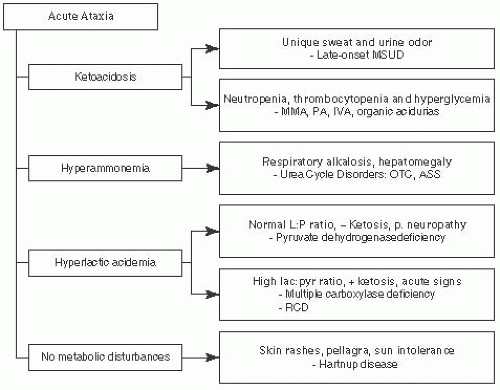 FIGURE 7.5 Acute Ataxia Associated with Metabolic Disorders. Adapted from Saudubray JM, Charpentier C. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York, NY: McGraw-Hill; 2000, with permission. |
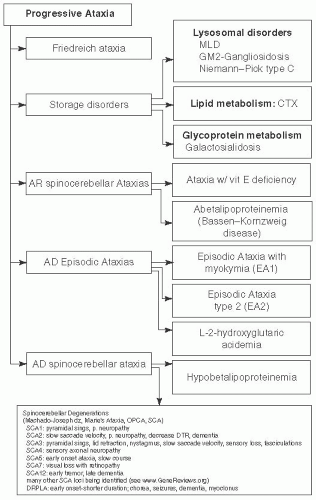 FIGURE 7.6 Progressive Ataxia Associated with Metabolic Disorders. Adapted from Saudubray JM, Charpentier C. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York, NY: McGraw-Hill; 2000, with permission. |
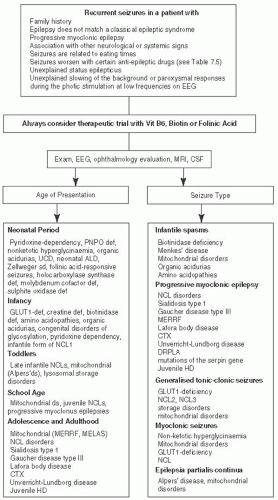 FIGURE 7.7 Epilepsies Associated with Metabolic Disorders. Adapted from Sedel F, Fontaine B, Saudubray JM, et al. Hereditary spastic paraparesis in adults associated with inborn errors of metabolism: a diagnostic approach. J Inherit Metab Dis. 2007;30(6):855-86450 and Wolf NC. Epilepsy in inborn errors of metabolism. Epileptic Disord. 2005; 7 (2): 67-81, with permission.51 |
TABLE 7.5 Anticonvulsants to be Avoided in Metabolic Disorders | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
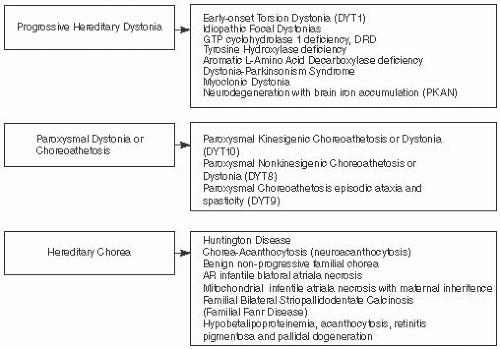 FIGURE 7.8 Movement Abnormalities Associated with Metabolic Disorders. |
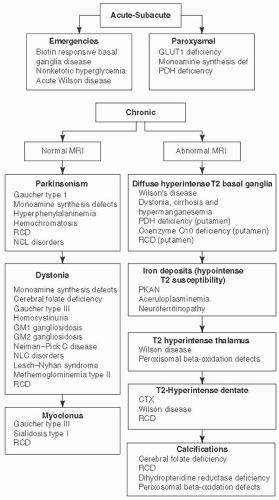 FIGURE 7.9 Movement Disorders Associated with Metabolic Disorders, Characterized by Onset and MRI Findings. Adapted from Sedel F, Saudubray JM, Roze E, et al. Movement disorders and inborn errors of metabolism in adults: a diagnostic approach. J Inherit Metab Dis. 2008;31(3):308-318, with permission.50 |
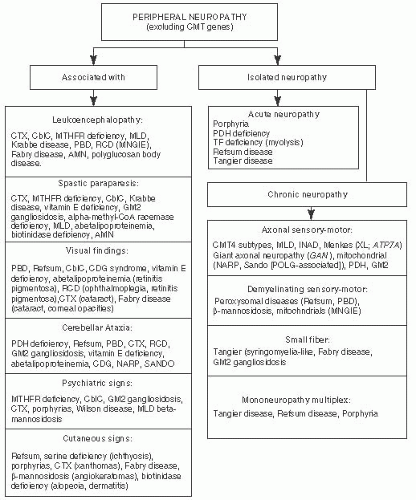 FIGURE 7.10 Peripheral Neuropathies Associated with Metabolic Disorders. Adapted from Sedel F, Baumann N, Turpin JC, et al. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis. 2007;30(5):631-641, with permission. |
TABLE 7.6 Ocular Findings Associated with Metabolic Disorders | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||
si/sx such as gait or speech disturbance, unchanged for years & then development of movement d/o (e.g., dysarthria, dystonia, dysdiadochokinesia, facio-linguo-pharyngeal rigidity, & gait difficulties). Other sx: rest & action tremor (rubral tremor) ± chorea & parkinsonism. Psychiatric si/sxprecedes movement d/o in 20% of cases.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


