40
CHAPTER
![]()
Metabolic Epilepsies
Abeer J. Hani and Mohamad A. Mikati
The spectrum of metabolic conditions causing epilepsy continues to expand, allowing for further identification of the etiologies of various epilepsies. Keeping track of these disorders can prove to be a daunting task for clinicians. A useful online resource in this regard is www.orpha.net that keeps a frequently updated database of the clinical manifestations of rare diseases, how to diagnose them, and of their potential treatments. In this chapter, a framework will be presented that permits clinicians to identify a patient with a possible metabolic epilepsy, assess the associated clinical features and neurophysiologic findings, and develop a diagnostic approach to such epilepsies. A brief overview of some of the more common metabolic epilepsies will be provided. For more details about the various inherited metabolic epilepsies, the reader is referred to more comprehensive reviews (1).
ETIOLOGIES
In many of the metabolic epilepsies that will be discussed in the further sections, occasional triggered seizures may occur in the setting of hypoglycemia or hyperammonemia. However, there is an intrinsic underlying epilepsy component in these conditions. It is thought that the pathogenesis of these conditions could be related to energy deficiency, toxic effects, impaired neuronal function, disturbance of neurotransmitter systems, associated brain malformations, vitamin or cofactor dependency, or vitamin transporter defects (Table 40.1) (2).
CLINICAL FEATURES
While such epilepsies often have unique clinical presentations, the following features may suggest a possible metabolic epilepsy. Onset is usually in the neonatal, infantile, or early childhood periods and rarely in adulthood (Table 40.2)(2,3). In the neonatal period, there may be reports of hypotonia, poor feeding, lethargy, respiratory distress, or lactic acidosis in combination with myoclonic seizures. In addition to myoclonic seizures, certain seizure types and epilepsy syndromes may be associated with specific metabolic epilepsies (Table 40.3) (4). Family history of similar presentations warrants further genetic and metabolic investigations. Refractoriness of the seizures to traditional antiepileptic drugs (AEDs) often raises a red flag in the neonatal and infantile age group for a possible metabolic etiology. A detailed history ought to be obtained to exclude other potential etiologies, including prenatal or perinatal events, head trauma, infections, or other systemic diseases. Physical examination should be detailed, focusing on features that can suggest other etiologies as well as those that could suggest metabolic etiologies. One should look for the presence of dysmorphic features or neurocutaneous stigmata, head size abnormalities, and various manifestations of systemic involvement. In addition to the seizures, other neurologic manifestations may be present and may predate or follow the seizures. These may include developmental delay/regression, intellectual disability, movement disorders, micro- or macrocephaly, and other cerebral gray or white matter changes.
TABLE 40.1 Pathogenesis of Common Metabolic Epilepsies
PATHOGENESIS | METABOLIC EPILEPSY |
Energy deficiency | Glucose transporter -1 (GLUT1) deficiency |
| Respiratory chain deficiency |
| Pyruvate dehydrogenase deficiency |
| Krebs cycle defects |
| Creatine deficiencies |
Toxic effects | Aminoacidopathies |
| Organic acidurias |
| Urea cycle defects |
| Molybdenum cofactor deficiency |
| Sulfite oxidase deficiency |
Impaired neuronal function | Storage diseases |
Disturbance of neurotransmitter system | Nonketotic hyperglycinemia |
| Atypical phenylketonuria |
| GABA transaminase deficiency |
| Succinic semi-aldehyde dehydrogenase deficiency |
Associated brain malformations | Peroxisomal disorders (Zellweger syndrome) |
| Respiratory chain deficiency |
| Pyruvate dehydrogenase deficiency |
| O-glycosylation defects (congenital muscular dystrophies) |
Vitamin or cofactor dependency and vitamin transporter defects | Biotinidase deficiency |
| Pyridoxine-dependent and pyridoxal 5’-phosphate- dependent epilepsy (folinic-acid-responsive seizures) |
| Thiamine transporter deficiency |
| Menkes’ disease |
| Folate transporter defect (FOLR1) |
| Dihydrofolate reductase deficiency |
Miscellaneous | Serine biosynthesis deficiency |
Source: Modified from Ref. (2). Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurol. 2014;13:727–739.
TABLE 40.2 Metabolic Epilepsies Stratified by Age at Manifestation
AGE AT MANIFESTATION | METABOLIC EPILEPSY |
Neonatal | – Pyridoxine-dependent epilepsy (including folinic-acid-responsive seizures) – Pyridox(am)ine 5-phosphate deficiency – Nonketotic hyperglycinemia – Urea cycle defects – Holocarboxylase synthase deficiency – Molybdenum cofactor deficiency – Sulfite oxidase deficiency – Organic acidurias – Zellweger syndrome – Neonatal adrenoleukodystrophy – Adenylosuccinate lyase deficiency – Dihydrofolate reductase deficiency |
Infancy | – Glucose transporter-1 (GLUT1) deficiency – Creatine deficiency – Biotinidase deficiency – Aminoacidopathies – Organic acidurias – Congenital disorders of glycosylation – Pyridoxine-dependent epilepsy – Infantile neuronal ceroid lipofuscinosis (CLN1) – Folate and thiamine transporter deficiencies – Peroxisomal disorders – Menkes disease |
Toddlers | – Late infantile neuronal ceroid lipofuscinosis (CLN2) – Mitochondrial disorders including Alpers disease – Lysosomal storage disorders – Thiamine transporter deficiency – Folate transporter deficiency |
School age and adolescence | – Mitochondrial disorders – Juvenile form of neuronal ceroid lipofuscinosis (CLN3) – Progressive myoclonic encephalopathies – Lysosomal storage disorders – Thiamine transporter deficiency – Lafora disease – Gaucher disease – Niemann-Pick type C disease |
Adulthood | – Kufs disease – Juvenile form of neuronal ceroid lipofuscinosis (CLN3) – Sialidosis type 1 – Gaucher disease – Mitchondrial diseases (MERRF, MELAS, NARP) – Nonsyndromic respiratory chain disorders – Lafora disease – Acute porphyrias – Wilson disease – GLUT1 deficiency – Creatine metabolism defects – SSADH deficiency – Urea cycle defects – Cerebrotendinous xanthomatosis – Metachromatic leukodystrophy – Adrenoleukodystrophy |
Source: Modified from Refs. (2) and (3).
Abbreviations: MELAS: mitochondrial encephalomyopathy, lactic acidosis, and stroke; MERRF: myoclonic epilepsy with ragged red fibers; NARP: neuropathy, ataxia, and retinitis pigmentosa; SSADH: succinic semialdehyde dehydrogenase.
SEIZURE TYPE/EPILEPSY SYNDROME | METABOLIC EPILEPSY |
Infantile spasms | – Biotinidase deficiency – Menkes disease – Mitochondrial disorders – Organic acidurias – Amino acidopathies |
Myoclonic seizures | – Nonketotic hyperglycinemia – Mitochondrial disorders – GLUT1 deficiency – Storage disorders |
Atypical absence | – GLUT1 deficiency |
Progressive myoclonic epilepsies | – Lafora disease? – Mitochondrial diseases (MERRF, MELAS) – Unverricht–Lundborg disease – Sialidosis |
Epilepsy with generalized tonic–clonic seizures | – GLUT1 deficiency? – Neuronal ceroid lipofuscinosis (NCL2/NCL3) – Other storage disorders – Mitochondrial disorders |
Epilepsia partialis continua | – Mitochondrial disorders with mutations in DNA polymerase gamma (Alpers disease). |
Status epilepticus | – Pyridoxine-dependent epilepsy – Alpers syndrome |
Source: Modified from Ref. (4). Bahi-Buisson N, Dulac O. Epilepsy in inborn errors of metabolism. Handb Clin Neurol. 2013;111:533–541.
ELECTROENCEPHALOGRAPHY
Given that multiple seizure types or epilepsy syndromes may be the presenting manifestations of metabolic epilepsies, an array of EEG presentations similarly exists (Figure 40.1). Although there are no pathognomonic EEG signatures of metabolic diseases, certain patterns may be associated with specific metabolic epilepsies (Table 40.4)(1).
In general, early-onset myoclonic metabolic epilepsy often manifests with burst suppression and/or irregular polyspike wave paroxysm during myoclonus. At times, hypsarrhythmia may be seen and rarely more specific patterns, such as the comb-like rhythm in maple syrup urine disease, may suggest a diagnosis. In progressive metabolic epilepsies, serial EEGs show progression from normal background to slowing, loss of sleep architecture, and an increased burden of generalized spike-and-wave activity.
NEUROIMAGING
Often the key features in the neuroimaging of this group of epilepsies include brain atrophy with symmetric findings. There may be myelination abnormalities with infrequent contrast enhancement (5). In certain metabolic epilepsies, there may be evidence of brain malformations, such as corpus callosal agenesis in glycine encephalopathy and pyruvate dehydrogenase deficiency or polymicrogyria in Zellweger syndrome. At times, predominant anatomic location of the white matter changes may suggest the diagnosis. White matter abnormalities are more anterior in Alexander disease, posterior in adrenoleukodystrophy, central in metachromatic leukodystrophy, diffuse in Pelizaeus-Merzbacher disease, and peripheral in L2-OH-glutaric aciduria (Figure 40.2).
It is useful to obtain neuroimaging for patients with metabolic epilepsies early in the course of their disease given that in later stages the findings are similar and consist of diffuse atrophy, reduced white matter volume, and shrunken basal ganglia (6). Often, the white matter is predominantly involved with or without involvement of the basal ganglia and thalami. The use of proton magnetic resonance spectroscopy (MRS) may assist in the diagnosis by demonstrating the presence of metabolites that are not normally detected, such as galactitol in galactosemia. Alternatively, MRS may show increased concentrations of metabolites as in Canavan disease or decreased concentrations as in creatine deficiency. Key radiologic features of some metabolic epilepsies are listed in Table 40.5 (7).
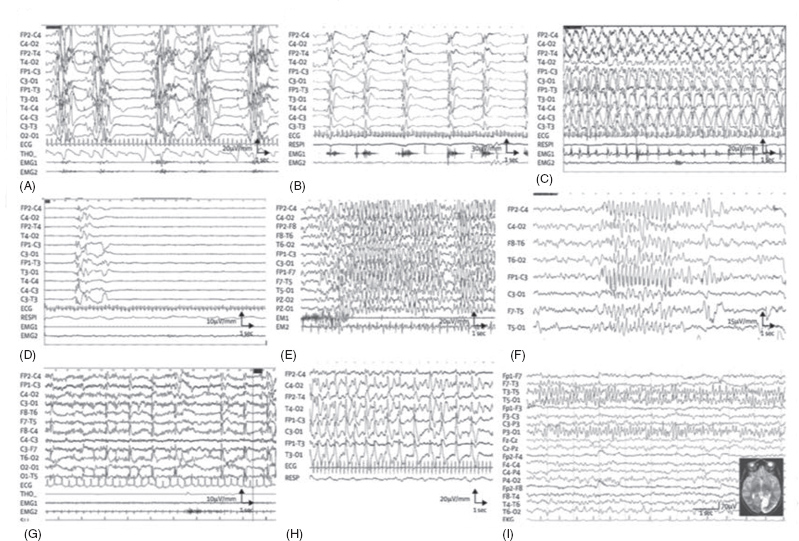
FIGURE 40.1 EEG in selected metabolic epilepsies. (A) Interictal EEG in a 15-day-old baby with nonketotic hyperglycinaemia, showing the characteristic suppression–bursts sequence (bursts of high-amplitude diffuse polyspikes separated by episodes of flat tracing). (B–D) EEG from a 3-month-old baby with pyridoxine-dependent epilepsy. (B) Epileptic spasms in clusters with high-amplitude slow complex and rhythmic waves. (C) High-amplitude rhythmic spike–waves predominate on the left hemisphere during a right clonic seizure. (D) After pyridoxine intravenous injection (100 mg), EEG shows flattening of the tracing. (E) Myoclonic status epilepticus in a 6-year-old girl with guanidinoacetate methyltransferase deficiency. Diffuse high-voltage spikes and spike–waves predominating on frontal or frontal-central areas; myoclonic jerks recorded on deltoid electromyography. (F) Atypical absence in an 11-year-old girl with glucose transporter-1 deficiency, with diffuse high-amplitude spike–waves. (G) Diffuse spikes, predominating on occipital areas triggered by slow photic stimulation in a 5-year-old girl with late-infantile neuronal ceroid lipofuscinosis. (H) A 3-year-old girl with Alpers disease due to POLG1 mutations. Interictal EEG shows bilateral high-amplitude periodic sharp waves and spike–waves with left occipital predominance. (I) A 16-year-old girl with MELAS and the classic mitochondrial 3243A→G mutation. EEG shows spike and spike–waves rhythmic discharges over the left posterior region, corresponding to the occipital area of hyperintense T2 signal. MELAS=mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes.
Source: Adapted from Ref. (2). Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurol. 2014;13:727–739.
![]()
COMMON METABOLIC EPILEPSIES
The spectrum of metabolic epilepsies is expanding. In this section, some of the more common and potentially treatable metabolic epilepsies will be detailed.
Vitamin B6-Dependent Epilepsies
Vitamin B6-dependent epilepsies are so named because vitamin B6 dependency is caused by a genetically inherited metabolic disorder requiring a lifelong need for pharmacologic doses of vitamin B6 and recurrence of seizures upon withdrawal (8). This is in contradistinction to vitamin B6 nutritional deficiency due to severe chronic malnutrition or vitamin B6 responsiveness seen in some epilepsy syndromes due to enhancement of the pyridoxine cofactor function without seizure recurrence upon withdrawal. Vitamin B6 is absorbed in three different vitamers (pyridoxal phosphate, pyridoxine, and pyridoxamine) and is then converted into pyridoxal-5’-phosphate (PLP) that acts as a cofactor in over 100 enzymatic reactions in amino acids and neurotransmitter metabolism.




