science revisited
The primary source of energy for resting muscle is derived from fatty acid oxidation (FAO). At rest, glucose utilization accounts for 10–15% of total oxygen consumption and both slow- and fast-twitch fibers have similar levels of glycogen content. The choice of bioenergetic pathway in working muscle depends on the type, intensity, and duration of exercise and also on diet and physical conditioning. In the first 5–10 min of moderate exercise, high-energy phosphates are used to first regenerate adenosine triphosphate (ATP). This is followed by muscle glycogen breakdown, which is indicated by a sharp rise in lactate during the first 10 min. Blood lactate levels then drop as muscle triglycerides and blood-borne fuels are used. After 90 min, the major fuels are glucose and free fatty acids (FFAs). During 1–4 h of mild-to-moderate prolonged exercise, muscle uptake of FFAs increases approximately 70% and, after 4 h, FFAs are used twice as much as carbohydrates.
Disorders of glycogen, lipid, or mitochondrial metabolism may cause two main clinical syndromes in muscle: (1) acute, recurrent, reversible muscle dysfunction with exercise intolerance and acute muscle breakdown or myoglobinuria (with or without cramps) and (2) progressive weakness. Progressive weakness and recurrent myoglobinuria can also occur together in a given disorder.
Clinical Syndrome of Myoglobinuria and Mechanisms
A common presentation for metabolic myopathies is recurrent myoglobinuria. Myoglobinuria is a clinical syndrome, not just a biochemical state.
 tips and tricks to diagnosis of myoglobinuria
tips and tricks to diagnosis of myoglobinuria
If the patient is alert, there is myalgia or limb weakness. The color of the urine is usually brownish, rather than red or pink, and the urine gives positive chemical tests for both albumin and heme, and there are few or no red blood cells. Myoglobin is identified by immunochemical methods in urine; however, serum creatine kinase (CK) is the most sensitive and readily available test to indicate muscle breakdown – CK is usually more than 100 times normal. Inconstant features include hyperuricemia, hyperphosphatemia, and hypo- or hypercalcemia. If there is renal failure, serum levels of potassium and calcium may rise. If the patient is comatose, or if the presenting disorder is one of acute renal failure, there may be no muscle symptoms or signs. The diagnosis can still be made if (1) the serum CK is 100 times normal and (2) there is renal failure. The main risks of an attack of myoglobinuria, which require careful monitoring, are potentially life-threatening respiratory failure, renal failure, and cardiac arrhythmias.
Etiologies of myoglobinuria can be divided into hereditary and sporadic forms. The sporadic etiologies that may precipitate an attack in an otherwise normal individual can be divided into those related to exertion, crush injury, ischemia, toxins and drugs, metabolic depression, abnormalities of body temperature, infections, progressive muscle disease, and those that appear to be idiopathic. A comparison of myoglobinuria related to exertion, heat stroke, neuroleptic malignant syndrome and malignant hyperthermia is given in Table 5.1.
Table 5.1. Heat, fever, and myoglobinuria
Reproduced from Rowland LP. Myoglobinuria. Can J Neurol Sci 1984;11:1–13 with permission from Canadian Journal Of Neurological Science.
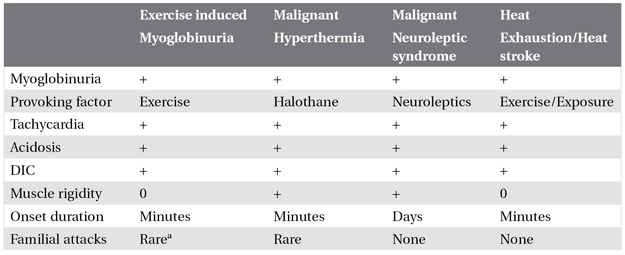
aHereditable biochemical abnormality may be identified.
DIC, disseminated intravascular coagulation.
The hereditary forms (Box 5.1) are particularly important because they are recurrent and suggest underlying pathogenic mechanisms. This may have implications for treatment strategies, preventive measures and genetic counseling. These forms may be divided into three groups based on whether the biochemical abnormality is known, incompletely characterized, or unknown. In the first group, there are at least 25 recognized disorders. All are autosomal recessive in inheritance with the exception of three X-linked disorders. The mitochondrial defects may be autosomal recessive, autosomal dominant, X-linked, sporadic, or inherited by maternal mitochondrial transmission.
Box 5.1. Heritable causes of exercise intolerance and recurrent myoglobinuria
Biochemical abnormality known
1. Glycolysis/Glycogenolysis
Phosphorylasea
Phosphofructokinase
Phosphoglycerate kinasea
Phosphoglycerate mutasea
Lactate dehydrogenasea
Phosphorylase “b” kinase
Debrancher
Aldolase Aa
2. Fatty acid oxidation
Carnitine palmitoyltransferase IIa
Long-chain acyl-CoA dehydrogenase
Very-long-chain acyl-CoA dehydrogenase
Medium-chain acyl-CoA dehydrogenase
Short-chain L-3-hydroxyacyl-CoA dehydrogenasea
Trifunctional protein/long-chain L-3-hydroxyacyl-CoA dehydrogenasea
Medium-chain 3-ketoacyl-CoA thiolasea
Acyl-CoA dehydrogenase 9 (ACAD9)a
3. Pentose phosphate pathway
Glucose-6-phosphate dehydrogenasea
4. Purine nucleotide cycle
Myoadenylate deaminase
5. Respiratory chain
Complex IIa and aconitase
Coenzyme Q
Multiple mitochondrial DNA deletions a
Complex Ia
Complex III (cytochrome b)
Complex IV (cytochrome oxidase)a
6. Triglyceride and membrane phospholipid biosynthesis
LPIN1 – muscle-specific phosphatidic acid phosphatasea
Biochemical abnormality incompletely characterized
Impaired long-chain fatty acid oxidationa
Impaired function of the sarcoplasmic reticulum in familial malignant hyperthermia (predisposition in central core disease)a
Abnormal composition of the sarcolemma in, for example, Duchenne and Becker muscular dystrophya
Biochemical abnormality unknown
Familial recurrent myoglobinuiraa
Repeated attacks in sporadic casesa
aEtiologies that have been documented to cause recurrent myoglobinuria beginning in childhood.
Modified from Tein I, DiMauro S, Rowland LP. Myoglobinuria. In: Rowland LP, DiMauro S (eds), Handbook of Clinical Neurology, vol 18. Myopathies. Amsterdam: Elsevier Science Publishers BV, 1992.
In FAO disorders, attacks of myoglobinuria are precipitated after mild-to-moderate prolonged exercise when fatty acids are the key energy source in exercising muscle. These attacks may be further exacerbated by inadequate caloric intake as in fasting or infection with vomiting, which further limit blood glucose. Other risk factors include infection, during which metabolic processes preferentially favor FAO; this persists despite glucose administration, thereby increasing the dependence on FAO. There may also be fever with shivering thermogenesis and vomiting with fasting, making this a common trigger. Cold exposure may be detrimental as shivering depends upon involuntary muscle activity which primarily depends upon long-chain fatty acids (LCFAs). Emotional stress has also been a recognized precipitant. Other possible mechanisms relate to the toxicity of elevated FFAs arising proximal to the block, especially LCFAs which may be membranotoxic.
Myoadenylate deaminase and glucose-6-phosphate dehydrogenase (G6PD) deficiencies have not been conclusively linked to causation because both enzymes may be absent in asymptomatic people.
There appear to be differences in the distribution of etiologies of heritable myoglobinuria in adults versus children. In a study of 100 cases of recurrent childhood-onset myoglobinuria, only 24% of children were diagnosed biochemically – 16 with carnitine palmitoyltransferase (CPT) II deficiency and 7 with various glycolytic/glycogenolytic defects. These children were divided into two groups: a type I exertional group with exertion as the primary precipitating factor and a type II toxic group with infection and/or fever and leukocytosis as the primary precipitants. The type II toxic childhood group was distinguished from the type I exertional childhood- and adult-onset groups by its etiologies, which were limited to FAO defects and slight female predominance in contrast to the marked male predominance in the latter two groups. The type II toxic group was further distinguished by its earlier age at onset of attacks of myoglobinuria, the presence of more generalized disease (e.g. ictal bulbar signs, encephalopathy, seizures, developmental delay), and a higher mortality rate. In a study of 77 adult patients, Tonin et al. identified the enzyme abnormality in 36 patients: CPT II deficiency in 17 patients; glycolytic/glycogenolytic defects in 15 patients; and combined CPT II and myoadenylate deaminase in one individual. The most common etiology overall for recurrent myoglobinuria in children, boys and girls, is CPT II deficiency and in adults is phosphorylase deficiency followed by CPT II deficiency. Differentiation of classic CPT II from phosphorylase deficiency is given in Table 5.2.
Table 5.2. Differentiation between disorders of glycogen versus lipid metabolism resulting in exercise intolerance and/or myoglobinuria
Taken with permission from Tein I. Approach to muscle cramps, exercise intolerance and recurrent myoglobinuria. Proceedings of the 38th Annual Meeting of the Canadian Congress of Neurosciences. Muscle Diseases Course, Quebec City, Canada, 2003: 1–29 (CME course).
| Glycolytic/Glycogenolytic | Fatty acid oxidation | |
| Phosphorylase deficiency | Carnitine palmitoyltransferase II deficiency “adult type” | |
| Symptom onset in exercise | Early (first few minutes) | Late (particularly after 1 hour) |
| Second wind | + | – |
| Myalgia | Cramps | Stiffness |
| Fixed weakness | More common | Less common |
| Elevated interictal creatine kinase | + | − |
| Abnormal forearm | + | − |
| ischemic lactate test | ||
| Delayed ketogenesis | − | + |
| Muscle biopsy | ± glycogen storage | ± lipid storage |
Glycolytic/Glycogenolytic Disorders
The clinical features, affected tissues, and enzyme defects of the muscle glycogenoses are shown in Table 5.3. Glycogen accumulation in muscle may or may not be present. Phosphorylase deficiency or McArdle’s disease is the most common glycogenosis resulting in recurrent myoglobinuria. Certain defects such as phosphorylase (PPL) and phosphofructokinase (PFK) deficiencies can be detected on muscle histochemical staining.
Table 5.3. Clinical presentation of muscle glycogenoses
Modified from DiMauro S, Lamperti C. Muscle glycogenoses. Muscle Nerve 2001;24:985.
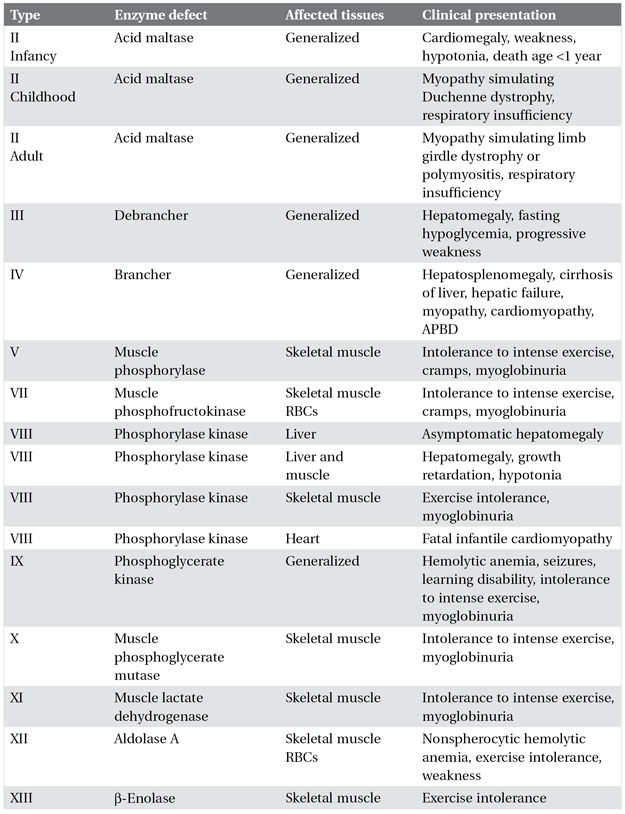
APBD, adult polyglucosan body disease; RBCs, red blood cells.
Individuals with defective glycolysis/glycogenolysis are most vulnerable during the initial stages of intense exercise, and they must rest soon after starting exercise because of muscle cramps. However, if they continue to exercise at low intensity for 10–12 min, they are then able to continue for a longer time. This is known as the second-wind phenomenon and has been attributed to a metabolic switch from carbohydrate to fatty acid utilization and to increased circulation, with increased availability of blood glucose from hepatic glycogenolysis.
The forearm ischemic exercise test is a useful test for the detection of enzymatic defects in the nonlysosomal glycogenolytic and glycolytic pathways.
 tips and tricks for performing forearm ischemic exercise test
tips and tricks for performing forearm ischemic exercise test
After 1 min of repetitive maximal grip exercise under ischemic conditions, blood samples from the antecubital vein are sequentially obtained at 1, 3, 5, 7, 10, and 15 min. In healthy individuals there is a four- to sixfold increase of lactate over baseline, with the peak occurring at 1–2 min after exercise, which declines to baseline values by 15 min. This is paralleled by a similar fivefold or more increase in ammonia, with levels generally peaking at 2–5 min after exercise in individuals with normal myoadenylate deaminase activity. In individuals with a defect in glycolysis/glycogenolysis, there is an insufficient rise in lactate (less than twofold), with a compensatory and exaggerated increase in ammonia, which also indicates sufficient effort on the part of the individual. An insufficient lactate rise has been demonstrated in PPL, debrancher, PFK, phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), and lactate dehydrogenase (LDH) deficiencies but not in acid maltase or phosphorylase b kinase deficiency. The major limitation of this test is that the rise of venous lactate in individuals who do not have a defect in this pathway is highly dependent on the patient’s ability and willingness to exercise. Therefore patients in whom lactate levels are low due to poor effort, or to placement of the venous line in other than the median cubital vein, show proportionally blunted ammonia responses. The test should be immediately truncated if the patient develops an acute cramp, because myonecrosis may occur in an individual with a glycolytic disorder.




